

Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options
- PMID: 32195193
- PMCID: PMC7064558
- DOI: 10.3389/fonc.2020.00298
Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options
Abstract
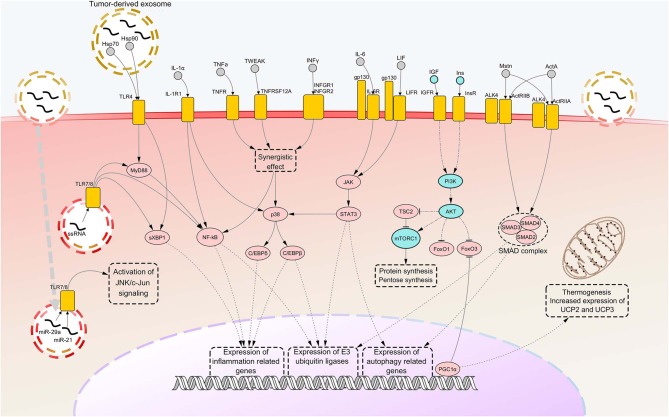
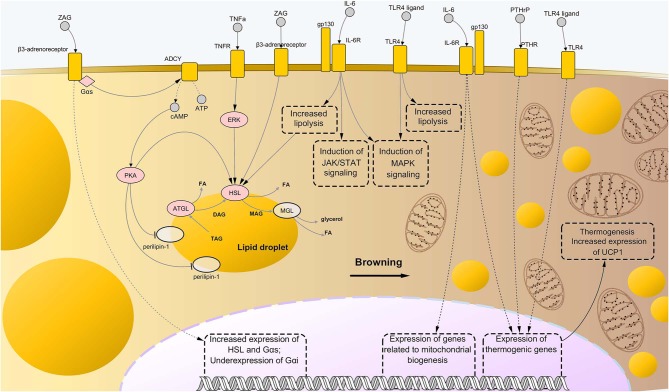
Cancer cachexia (CC) is a multifactorial syndrome characterized by systemic inflammation, uncontrolled weight loss and dramatic metabolic alterations. This includes myofibrillar protein breakdown, increased lipolysis, insulin resistance, elevated energy expediture, and reduced food intake, hence impairing the patient's response to anti-cancer therapies and quality of life. While a decade ago the syndrome was considered incurable, over the most recent years much efforts have been put into the study of such disease, leading to the development of potential therapeutic strategies. Several important improvements have been reached in the management of CC from both the diagnostic-prognostic and the pharmacological viewpoint. However, given the heterogeneity of the disease, it is impossible to rely only on single variables to properly treat patients presenting this metabolic syndrome. Moreover, the cachexia symptoms are strictly dependent on the type of tumor, stage and the specific patient's response to cancer therapy. Thus, the attempt to translate experimentally effective therapies into the clinical practice results in a great challenge. For this reason, it is of crucial importance to further improve our understanding on the interplay of molecular mechanisms implicated in the onset and progression of CC, giving the opportunity to develop new effective, safe pharmacological treatments. In this review we outline the recent knowledge regarding cachexia mediators and pathways involved in skeletal muscle (SM) and adipose tissue (AT) loss, mainly from the experimental cachexia standpoint, then retracing the unimodal treatment options that have been developed to the present day.
Keywords: animal models; cachexia mediators; cancer cachexia; clinical trials; muscle tissue and adipose tissue loss; therapeutic strategies.
Copyright © 2020 Marceca, Londhe and Calore.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources