

Biased GPCR signaling: Possible mechanisms and inherent limitations
- PMID: 32201315
- PMCID: PMC7275904
- DOI: 10.1016/j.pharmthera.2020.107540
Biased GPCR signaling: Possible mechanisms and inherent limitations
Erratum in
-
Corrigendum to "Biased GPCR signaling: Possible mechanisms and inherent limitations" [Pharmacology & Therapeutics 211 (2020) 107540].Pharmacol Ther. 2020 Sep;213:107615. doi: 10.1016/j.pharmthera.2020.107615. Epub 2020 Jul 2. Pharmacol Ther. 2020. PMID: 32622639 No abstract available.
Abstract
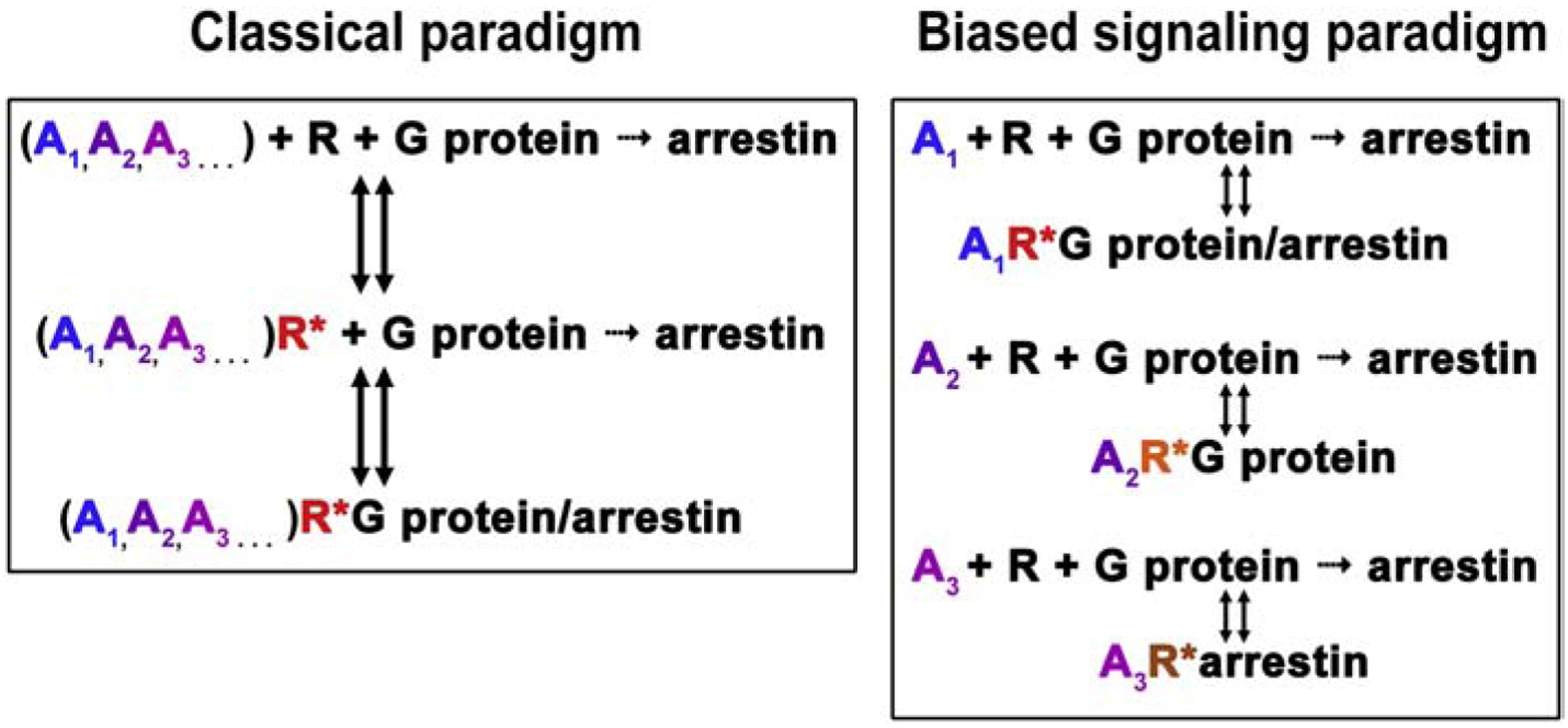
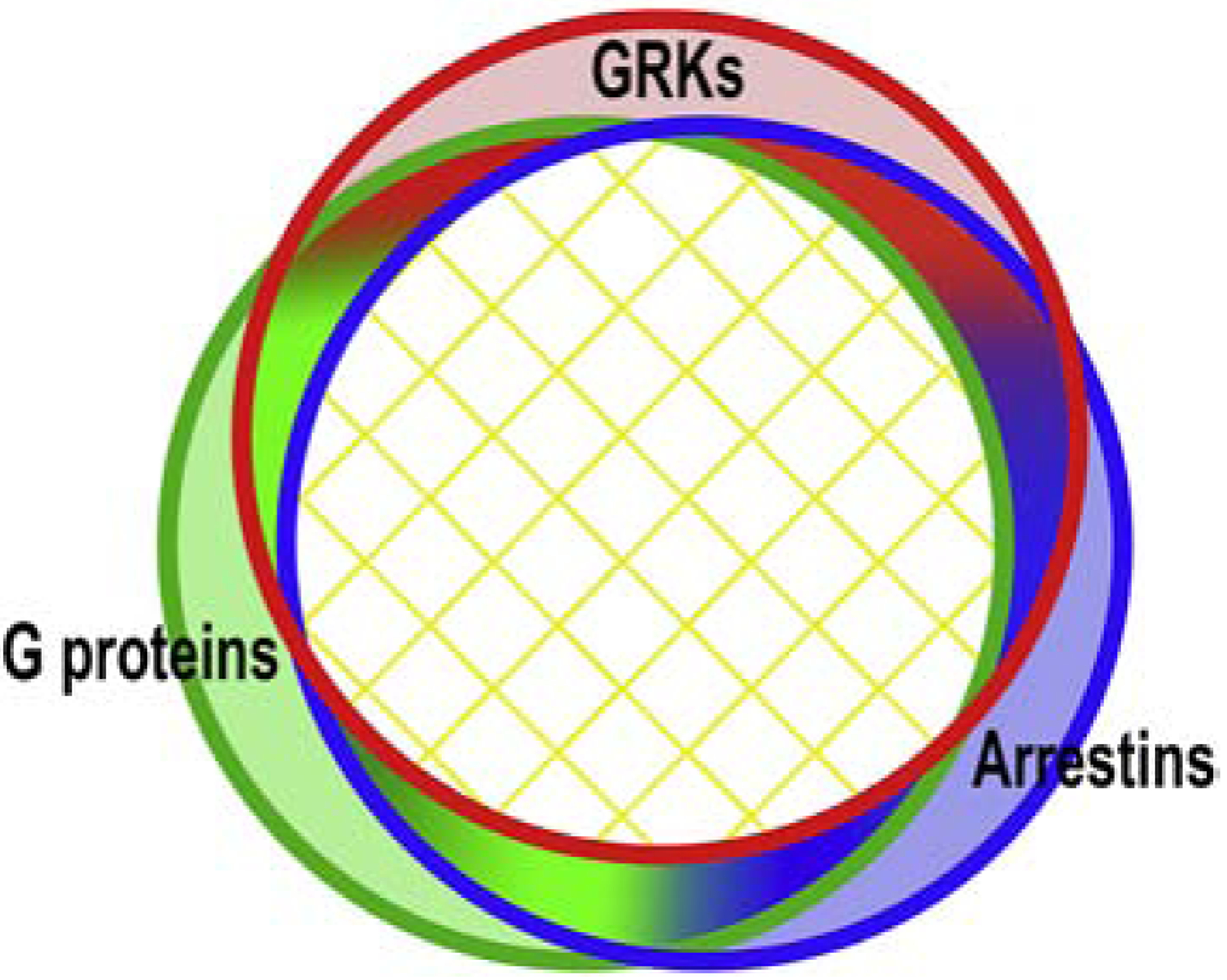
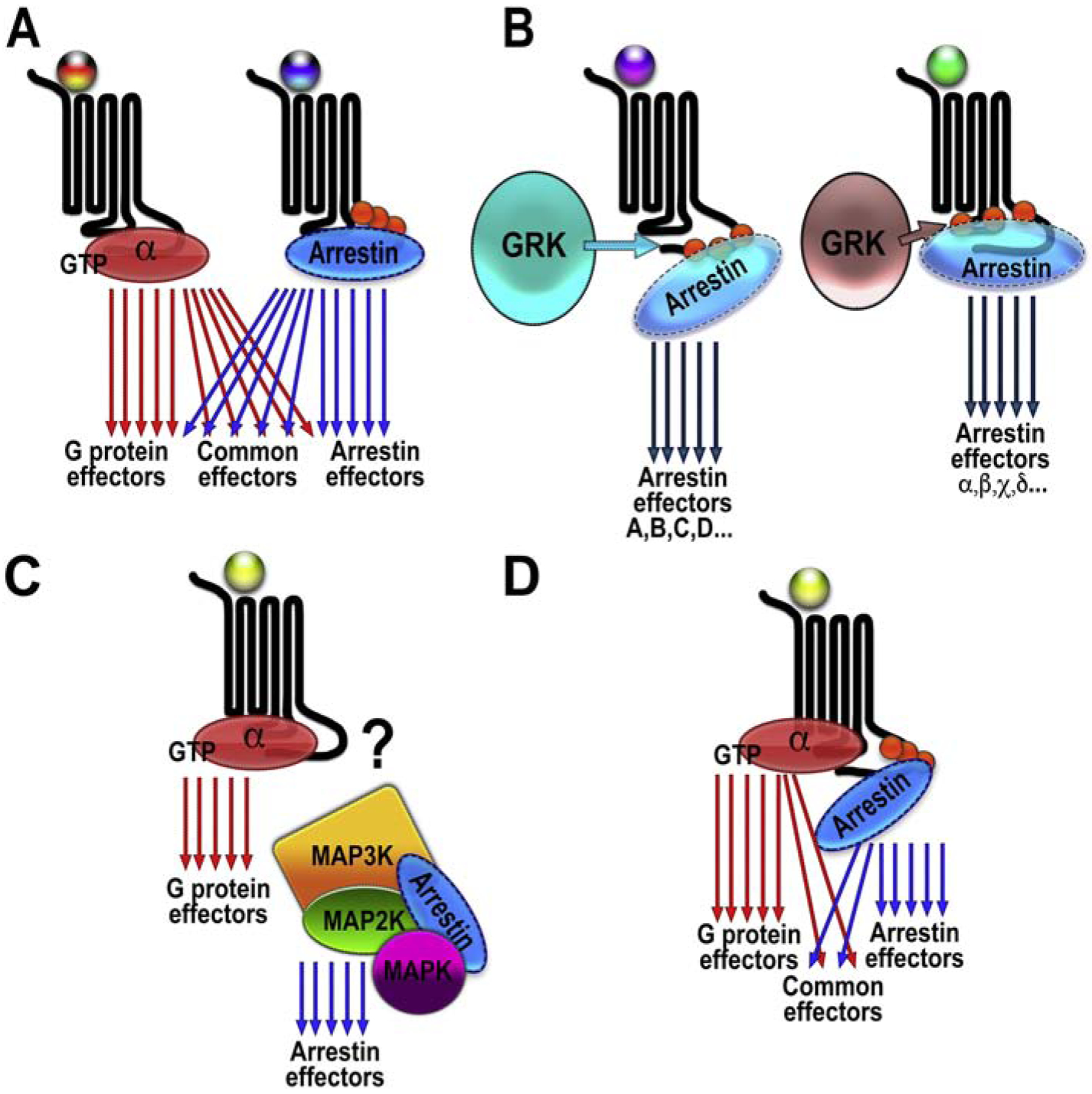
G protein-coupled receptors (GPCRs) are targeted by about a third of clinically used drugs. Many GPCRs couple to more than one type of heterotrimeric G proteins, become phosphorylated by any of several different GRKs, and then bind one or more types of arrestin. Thus, classical therapeutically active drugs simultaneously initiate several branches of signaling, some of which are beneficial, whereas others result in unwanted on-target side effects. The development of novel compounds to selectively channel the signaling into the desired direction has the potential to become a breakthrough in health care. However, there are natural and technological hurdles that must be overcome. The fact that most GPCRs are subject to homologous desensitization, where the active receptor couples to G proteins, is phosphorylated by GRKs, and then binds arrestins, suggest that in most cases the GPCR conformations that facilitate their interactions with these three classes of binding partners significantly overlap. Thus, while partner-specific conformations might exist, they are likely low-probability states. GPCRs are inherently flexible, which suggests that complete bias is highly unlikely to be feasible: in the conformational ensemble induced by any ligand, there would be some conformations facilitating receptor coupling to unwanted partners. Things are further complicated by the fact that virtually every cell expresses numerous G proteins, several GRK subtypes, and two non-visual arrestins with distinct signaling capabilities. Finally, novel screening methods for measuring ligand bias must be devised, as the existing methods are not specific for one particular branch of signaling.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare no conflict of interest.
Figures
References
-
- Ahn S, Shenoy SK, Wei H, & Lefkowitz RJ (2004). Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem, 279, 35518–35525. - PubMed
-
- Ahn S, Wei H, Garrison TR, & Lefkowitz RJ (2004). Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by beta-arrestins 1 and 2. J Biol Chem, 279, 7807–7811. - PubMed
-
- Amarandi R-M, Hjortø GM, Rosenkilde MM, & Karlshøj S (2016). Chapter Eight - Probing Biased Signaling in Chemokine Receptors In Handel TM (Ed.), Methods in Enzymology (Vol. 570, pp. 155–186): Academic Press. - PubMed
-
- Anthony DF, Sin YY, Vadrevu S, Advant N, Day JP, Byrne AM, Lynch MJ, Milligan G, Houslay MD, & Baillie GS (2011). β-Arrestin 1 inhibits the GTPase-activating protein function of ARHGAP21, promoting activation of RhoA following angiotensin II type 1A receptor stimulation. Mol Cell Biol, 31, 1066–1075. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
