

AD "Statin": Alzheimer's Disorder is a "Fast" Disease Preventable by Therapeutic Intervention Initiated Even Late in Life and Reversible at the Early Stages
- PMID: 32201863
- PMCID: PMC7083596
- DOI: 10.33597/aimm.02-1006
AD "Statin": Alzheimer's Disorder is a "Fast" Disease Preventable by Therapeutic Intervention Initiated Even Late in Life and Reversible at the Early Stages
Abstract
The present study posits that Alzheimer's disorder is a "fast" disease. This is in sharp contrast to a view, prevailing until now, that Alzheimer's Disease (AD) is a quintessential "slow" disease that develops throughout the life as one prolonged process. According to this view, beta-amyloid (Aβ) is produced and secreted solely by the beta-amyloid precursor protein (βAPP) proteolytic/secretory pathway. As its extracellular levels increase, it triggers neurodegeneration starting relatively early in life. Damages accumulate and manifest, late in life in sporadic Alzheimer's Disease (SAD) cases, as AD symptoms. In familial AD (FAD) cases, where mutations in βAPP gene or in presenilins increase production of either common Aβ isoform or of its more toxic isoforms, neurodegeneration reaches critical threshold sooner and AD symptoms occur earlier in life, mostly in late 40s and 50s. There are currently no preventive AD therapies but if they were available, according to this viewpoint it would be largely futile to intervene late in life in case of potential SAD or at mid-age in cases of FAD because, although AD symptoms have not yet manifested, the damage has already occurred during the preceding decades. In this paradigm, to be effective, preventive therapeutic intervention should be initiated early in life. The outlook suggested by the present study is radically different. According to it, Alzheimer's disease evolves in two stages. The first stage is a slow process of intracellular beta-amyloid accumulation. It occurs via βAPP proteolytic/secretory pathway and cellular uptake of secreted Aβ common to Homo sapiens, including healthy humans, and to non-human mammals, and results neither in significant damage, nor in manifestation of the disease. The second stage occurs exclusively in humans, commences shortly before symptomatic onset of the disease, sharply accelerates the production and increases intracellular levels of Aβ that is not secreted but is retained intracellularly, generates significant damages, triggers AD symptoms, and is fast. It is driven by an Aβ generation pathway qualitatively and quantitatively different from βAPP proteolytic process and entirely independent of beta-amyloid precursor protein, and results in rapid and substantial intracellular accumulation of Aβ, consequent significant neurodegeneration, and symptomatic AD. In this paradigm, a preventive therapy for AD, an AD "statin", would be effective when initiated at any time prior to commencement of the second stage. Moreover, there are good reasons to believe that with a drug blocking βAPP-independent Aβ production pathway in the second stage, it would be possible not only to preempt the disease but also to stop and to reverse it even when early AD symptoms have already manifested. The present study posits a notion of AD as a Fast Disease, offers evidence for the occurrence of the AD-specific Aβ production pathway, describes cellular and molecular processes constituting an engine that drives Alzheimer's disease, and explains why non-human mammals are not susceptible to AD and why only a subset of humans develop the disease. It establishes that Alzheimer's disease is preventable by therapeutic intervention initiated even late in life, details a powerful mechanism underlying the disease, suggests that Aβ produced in the βAPP-independent pathway is retained intracellularly, elaborates why neither BACE inhibition nor Aβ immunotherapy are effective in treatment of AD and why intracellularly retained beta-amyloid could be the primary agent of neuronal death in Alzheimer's disease, necessitates generation of a novel animal AD model capable of producing Aβ via βAPP-independent pathway, proposes therapeutic targets profoundly different from previously pursued components of the βAPP proteolytic pathway, and provides conceptual rationale for design of drugs that could be used not only preemptively but also for treatment and reversal of the early stages of the disease.
Keywords: Alzheimer’s disease; Asymmetric RNA-dependent beta-APP mRNA amplification; Mitochondrial dysfunction-related stresses; Precursor-independent generation of beta-amyloid.
Conflict of interest statement
Conflict of Interest Authors declare no conflict of interest.
Figures
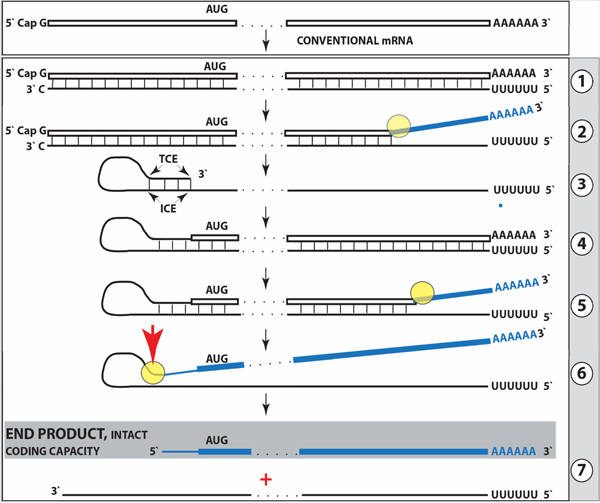
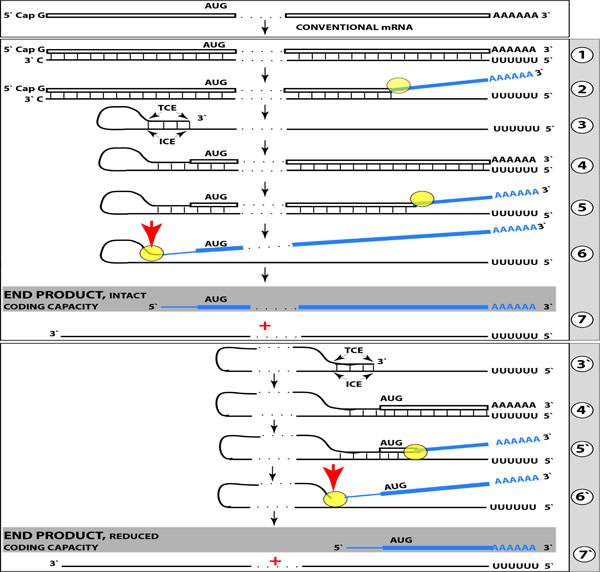
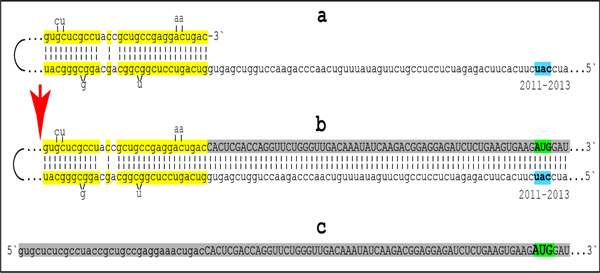


References
-
- Haass C, Lemere C, Capell A, Citron M, Seubert P, Schenk D, et al. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1(12):1291–6. - PubMed
-
- Dyrks T, Dyrks E, Monning U, Urmoneit B, Turner J, Beyreuther, K. Generation of beta A4 from the amyloid protein precursor and fragments thereof. FEBS Lett. 1993;335(1):89–93. - PubMed
-
- Iizuka T, Shoji M, Kawarabayashi T, Sato M, Kobayashi T, Tada N, et al. Intracellular generation of amyloid beta-protein from amyloid beta-protein precursor fragment by direct cleavage with beta- and gamma-secretase. Biochem Biophys Res Commun. 1996;218(1):238–42. - PubMed
-
- DeStrooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113(11):1857–70. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials