

Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms
- PMID: 32201880
- PMCID: PMC7441741
- DOI: 10.1210/endrev/bnaa006
Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms
Abstract
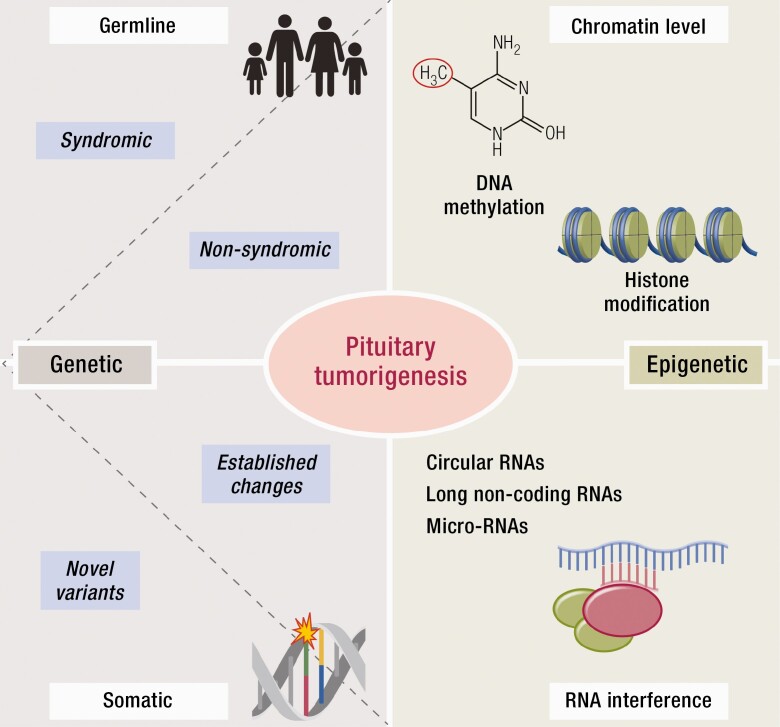
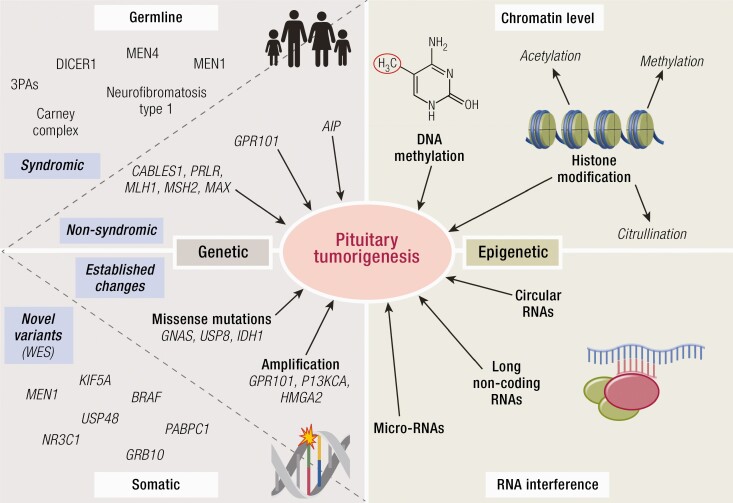
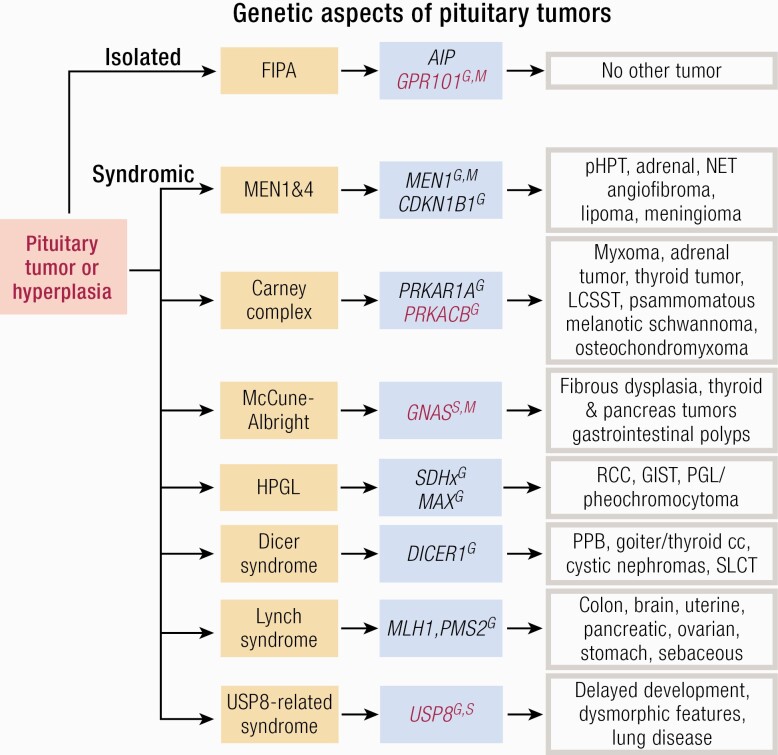
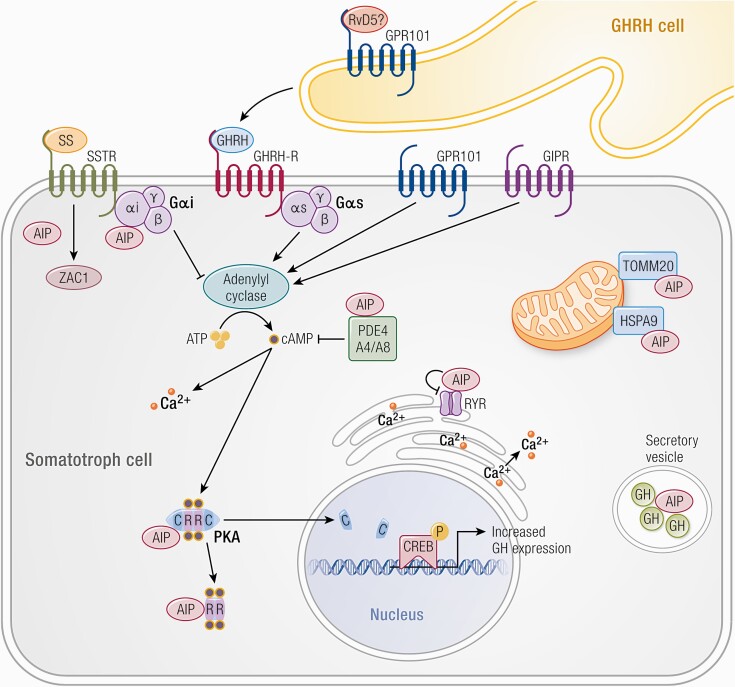
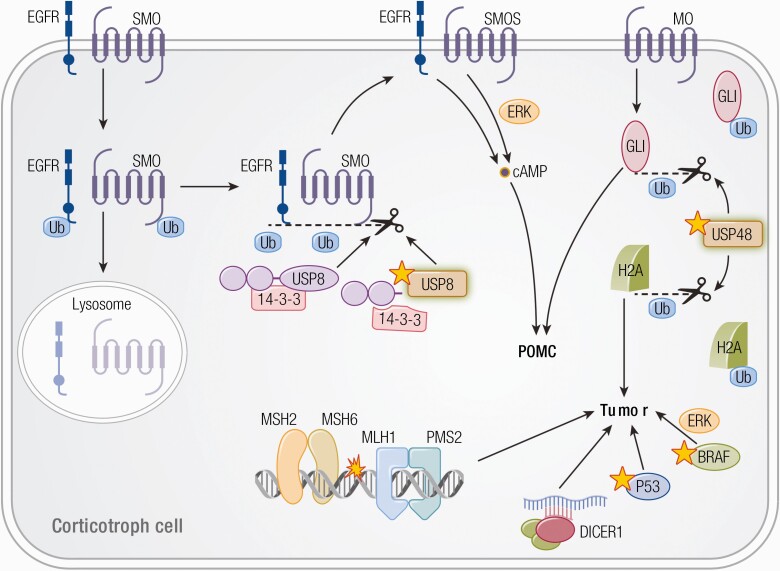
Substantial advances have been made recently in the pathobiology of pituitary tumors. Similar to many other endocrine tumors, over the last few years we have recognized the role of germline and somatic mutations in a number of syndromic or nonsyndromic conditions with pituitary tumor predisposition. These include the identification of novel germline variants in patients with familial or simplex pituitary tumors and establishment of novel somatic variants identified through next generation sequencing. Advanced techniques have allowed the exploration of epigenetic mechanisms mediated through DNA methylation, histone modifications and noncoding RNAs, such as microRNA, long noncoding RNAs and circular RNAs. These mechanisms can influence tumor formation, growth, and invasion. While genetic and epigenetic mechanisms often disrupt similar pathways, such as cell cycle regulation, in pituitary tumors there is little overlap between genes altered by germline, somatic, and epigenetic mechanisms. The interplay between these complex mechanisms driving tumorigenesis are best studied in the emerging multiomics studies. Here, we summarize insights from the recent developments in the regulation of pituitary tumorigenesis.
Keywords: PitNET; pituitary adenoma; pituitary neoplasm; pituitary tumor; pituitary tumorigenesis.
© Endocrine Society 2020.
Figures
References
-
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101(3):613–619. - PubMed
-
- Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006;91(12):4769–4775. - PubMed
-
- Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72(3):377–382. - PubMed
-
- Fontana E, Gaillard R. [Epidemiology of pituitary adenoma: results of the first Swiss study]. Rev Med Suisse. 2009;5(223):2172–2174. - PubMed
-
- Raappana A, Koivukangas J, Ebeling T, Pirila T. Incidence of pituitary adenomas in Northern Finland in 1992–2007. J Clin Endocrinol Metab. 2010;95(9):4268–4275. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
