

Simulation-Based Methods for Model Building and Refinement in Cryoelectron Microscopy
- PMID: 32202798
- PMCID: PMC7284120
- DOI: 10.1021/acs.jcim.0c00087
Simulation-Based Methods for Model Building and Refinement in Cryoelectron Microscopy
Abstract
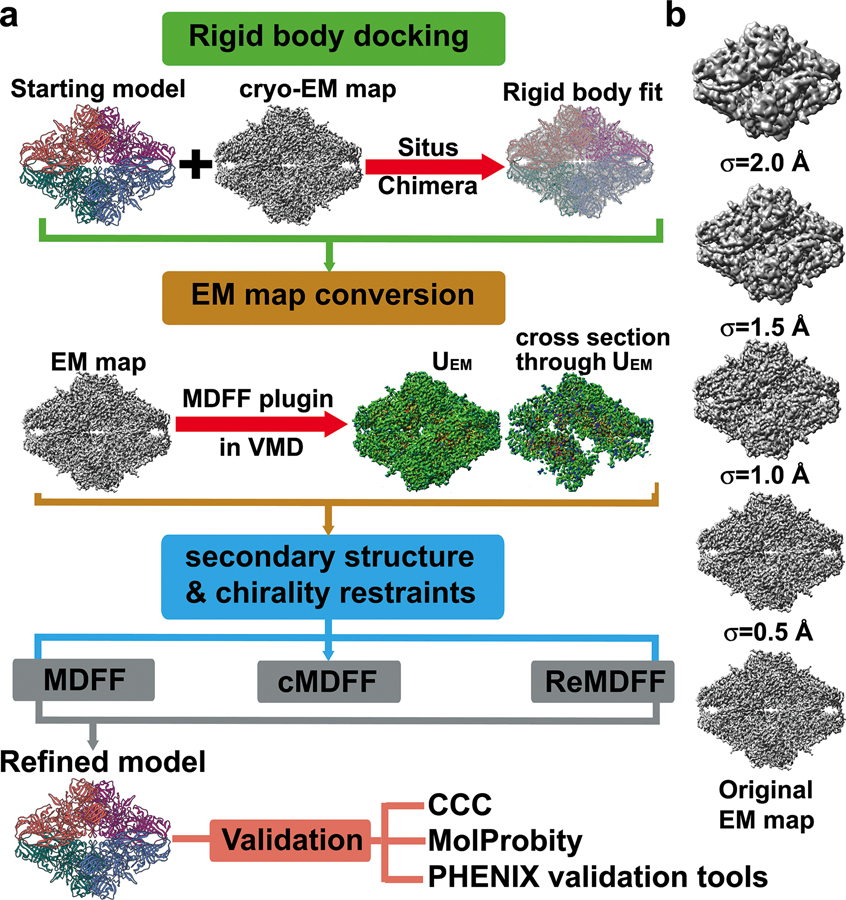
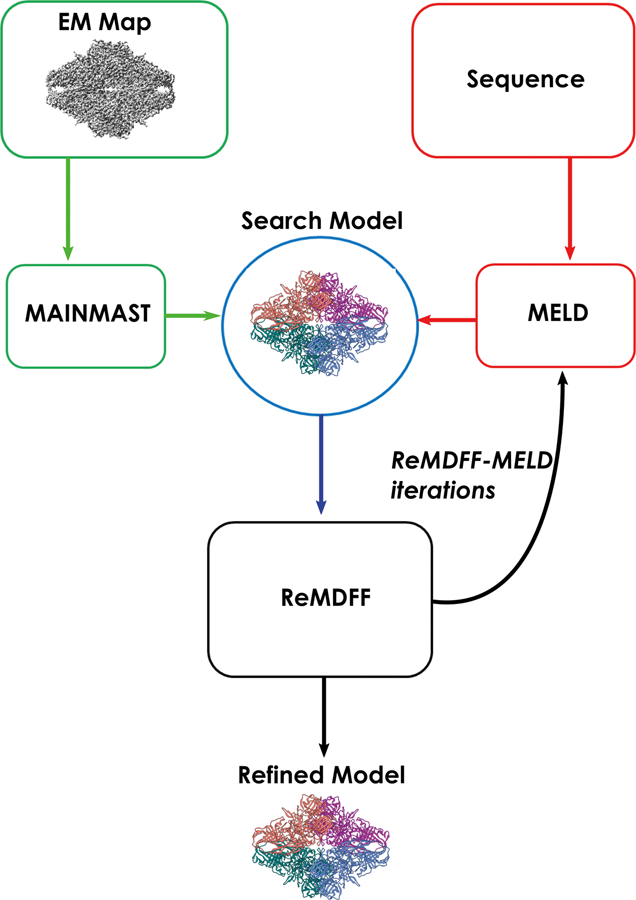
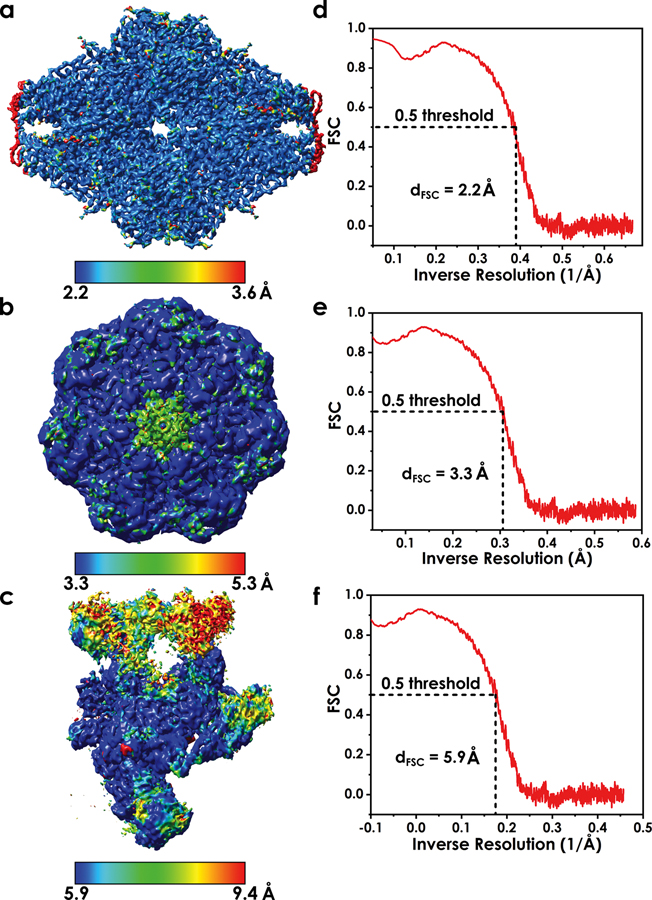
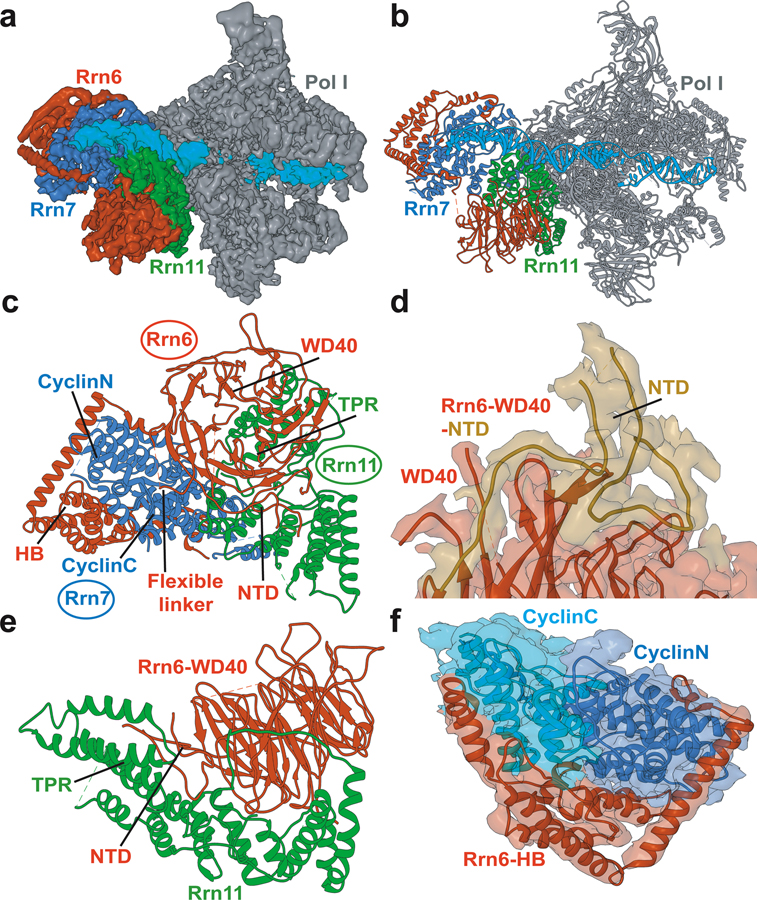
Advances in cryoelectron microscopy (cryo-EM) have revolutionized the structural investigation of large macromolecular assemblies. In this review, we first provide a broad overview of modeling methods used for flexible fitting of molecular models into cryo-EM density maps. We give special attention to approaches rooted in molecular simulations-atomistic molecular dynamics and Monte Carlo. Concise descriptions of the methods are given along with discussion of their advantages, limitations, and most popular alternatives. We also describe recent extensions of the widely used molecular dynamics flexible fitting (MDFF) method and discuss how different model-building techniques could be incorporated into new hybrid modeling schemes and simulation workflows. Finally, we provide two illustrative examples of model-building and refinement strategies employing MDFF, cascade MDFF, and RosettaCM. These examples come from recent cryo-EM studies that elucidated transcription preinitiation complexes and shed light on the functional roles of these assemblies in gene expression and gene regulation.
Keywords: RNA polymerases; cryoelectron microscopy; de novo model building; gene regulation; hybrid methods; molecular dynamics flexible fitting (MDFF); molecular modeling; transcription preinitiation complexes.
Figures
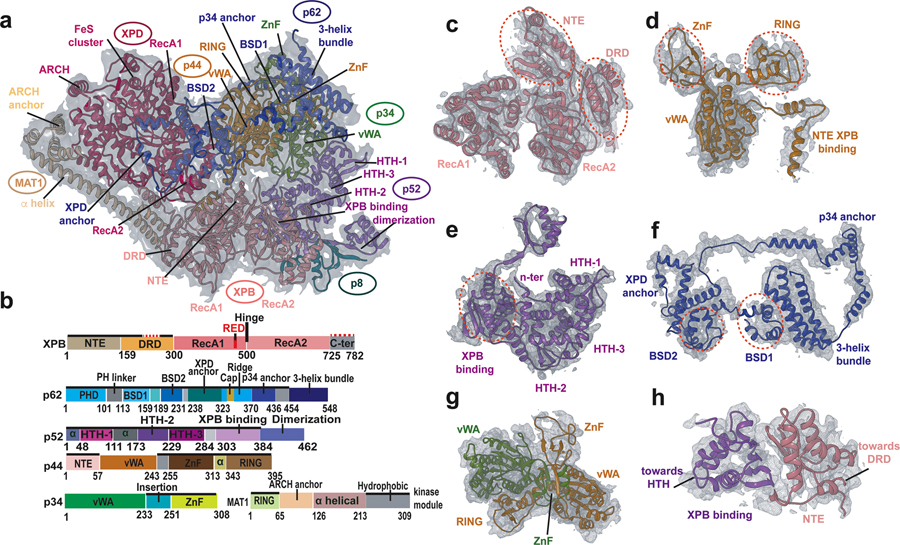
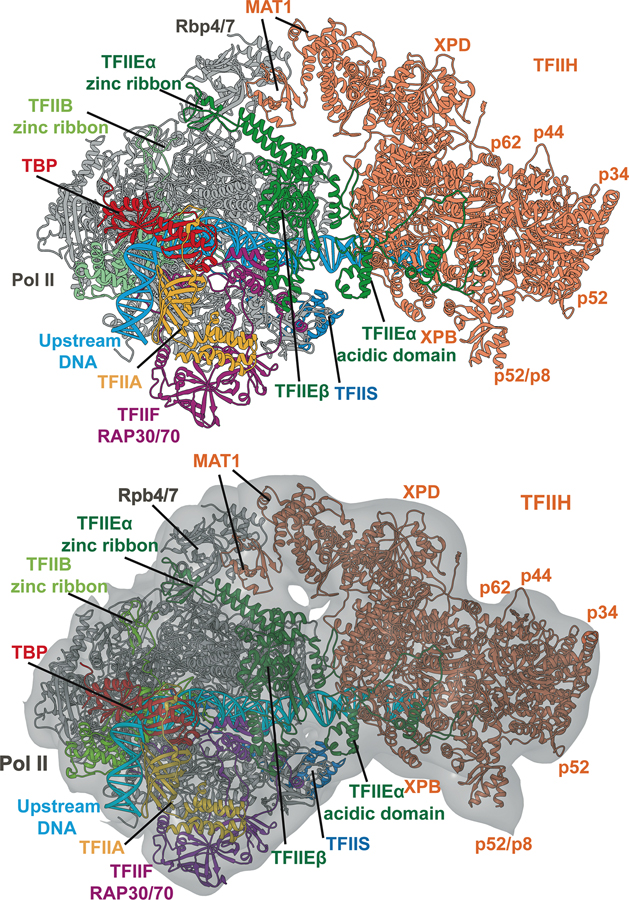
References
-
- Bai XC; McMullan G; Scheres SH How cryo-EM is revolutionizing structural biology. Trends Biochem Sci 2015, 40 (1), 49–57. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
