

DNA methylation disruption reshapes the hematopoietic differentiation landscape
- PMID: 32203468
- PMCID: PMC7216752
- DOI: 10.1038/s41588-020-0595-4
DNA methylation disruption reshapes the hematopoietic differentiation landscape
Abstract
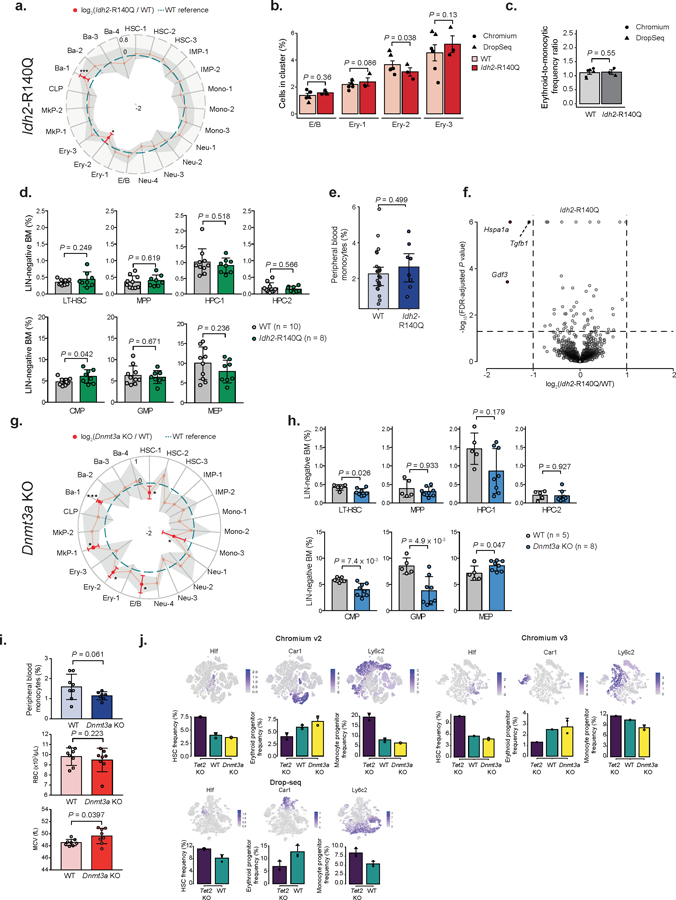
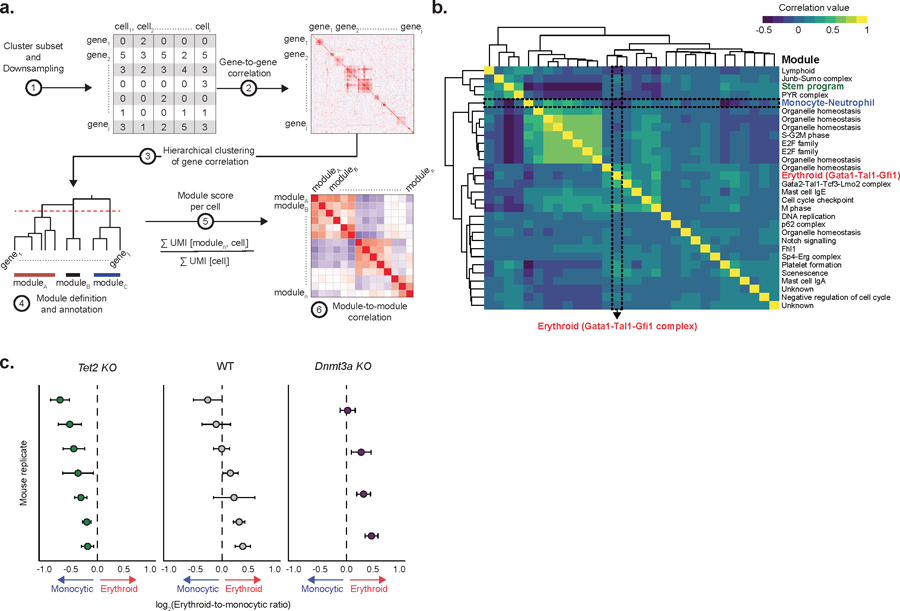
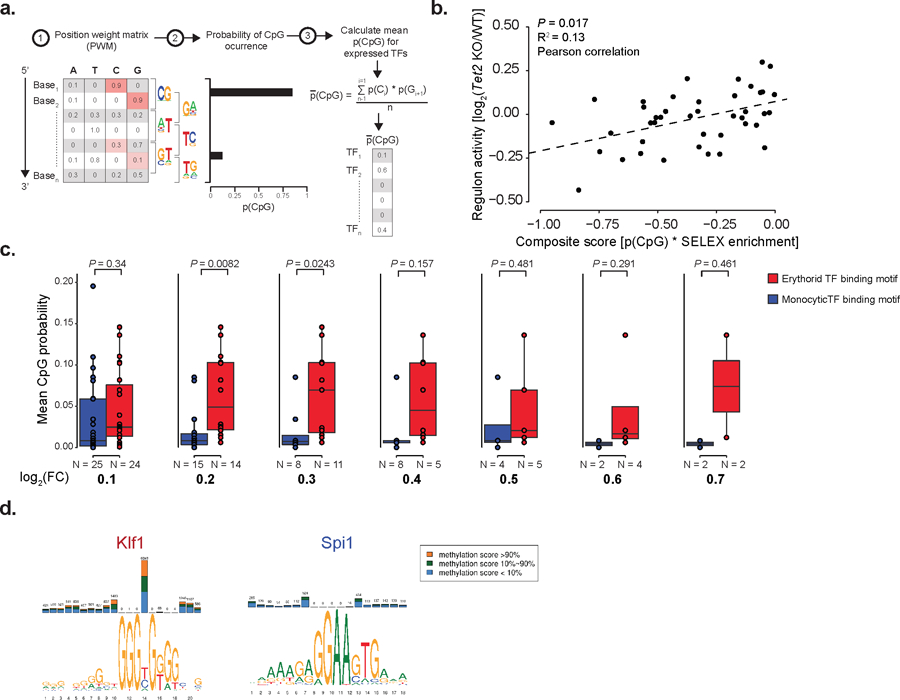
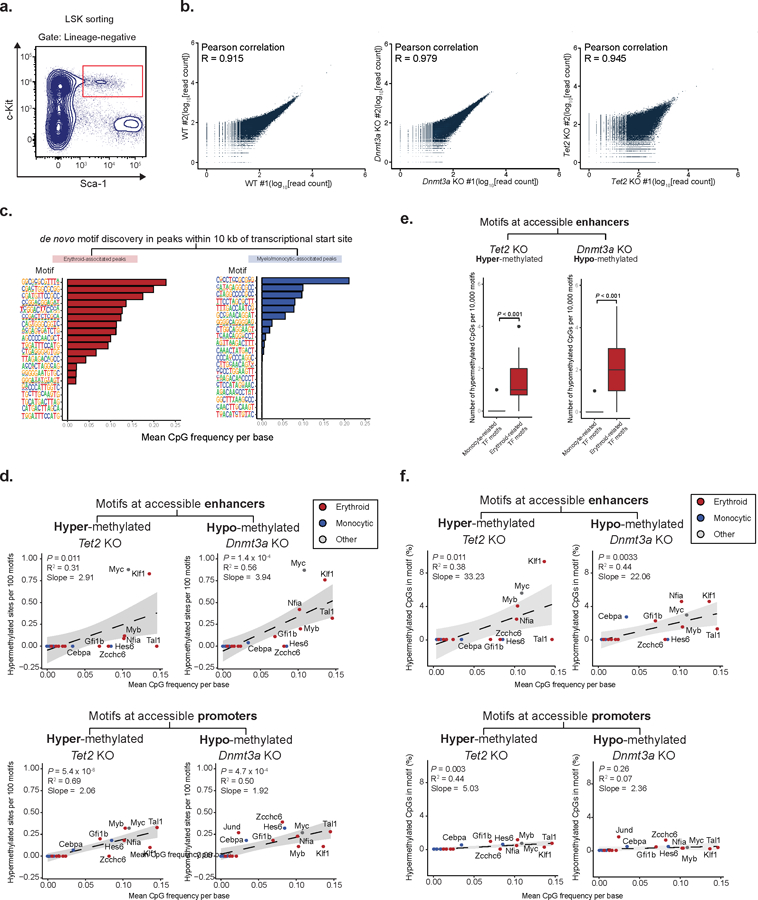
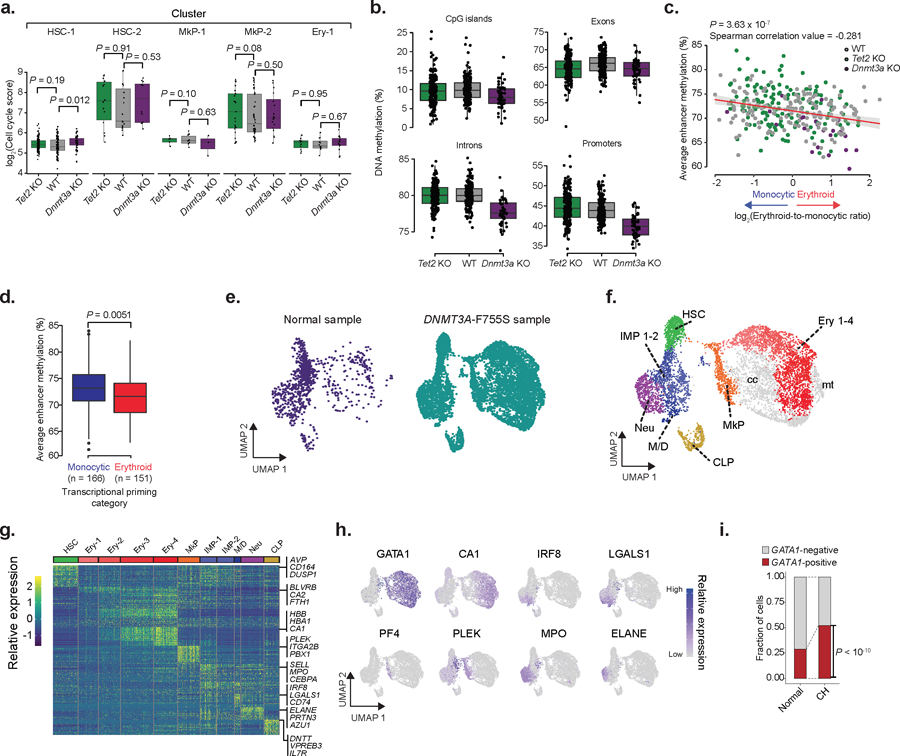
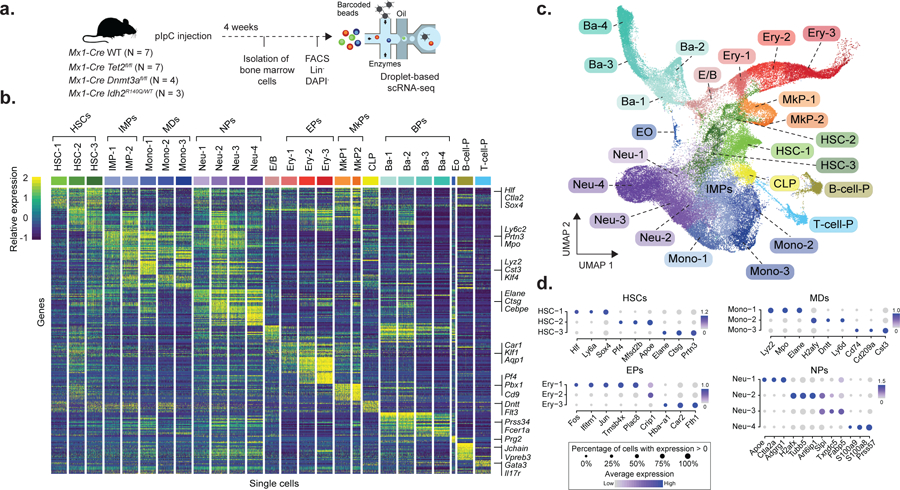
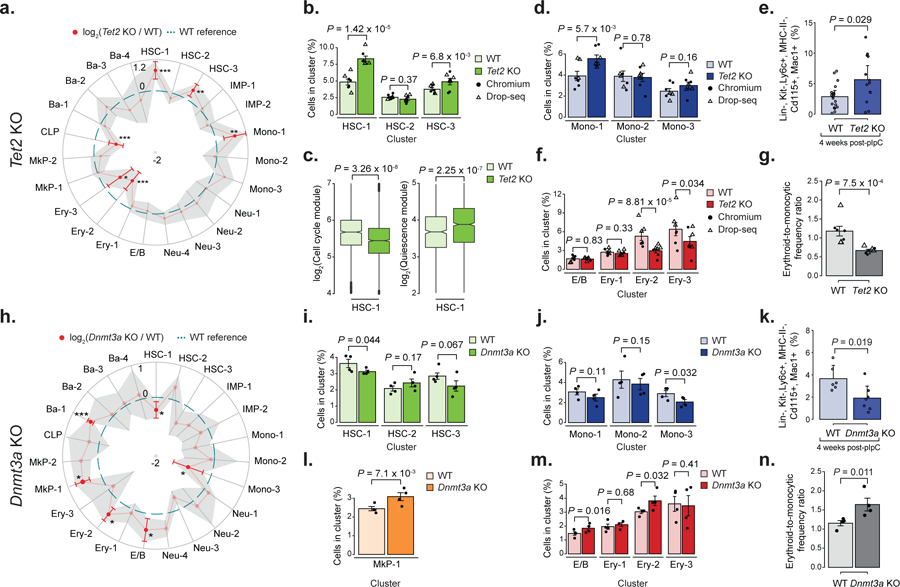
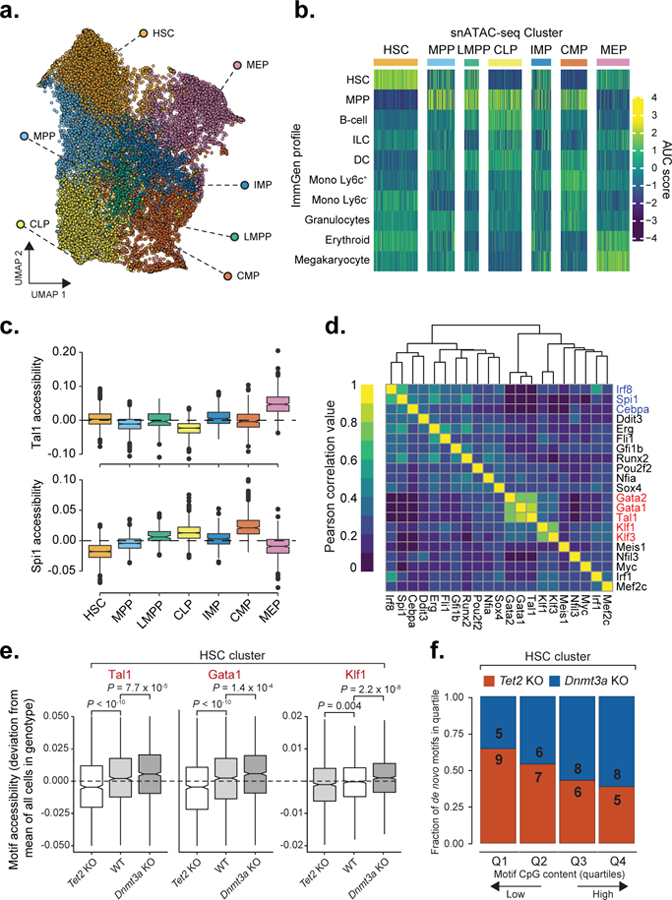
Mutations in genes involved in DNA methylation (DNAme; for example, TET2 and DNMT3A) are frequently observed in hematological malignancies1-3 and clonal hematopoiesis4,5. Applying single-cell sequencing to murine hematopoietic stem and progenitor cells, we observed that these mutations disrupt hematopoietic differentiation, causing opposite shifts in the frequencies of erythroid versus myelomonocytic progenitors following Tet2 or Dnmt3a loss. Notably, these shifts trace back to transcriptional priming skews in uncommitted hematopoietic stem cells. To reconcile genome-wide DNAme changes with specific erythroid versus myelomonocytic skews, we provide evidence in support of differential sensitivity of transcription factors due to biases in CpG enrichment in their binding motif. Single-cell transcriptomes with targeted genotyping showed similar skews in transcriptional priming of DNMT3A-mutated human clonal hematopoiesis bone marrow progenitors. These data show that DNAme shapes the topography of hematopoietic differentiation, and support a model in which genome-wide methylation changes are transduced to differentiation skews through biases in CpG enrichment of the transcription factor binding motif.
Conflict of interest statement
Competing interests
OA-W has served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, and Janssen, and is on the Scientific Advisory Board of Envisagenics Inc; OA-W has received prior research funding from H3B Biomedicine unrelated to the current manuscript. R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Loxo (until 2/2019), Imago, C4 Therapeutics and Isoplexis, which each include an equity interest. He receives research support from and consulted for Celgene and Roche, he has received research support from Prelude Therapeutics, and he has consulted for Lilly, Incyte, Novartis, Astellas, Morphosys and Janssen. He has received honoraria from Lilly and Amgen for invited lectures and from Gilead for grant reviews.
Figures

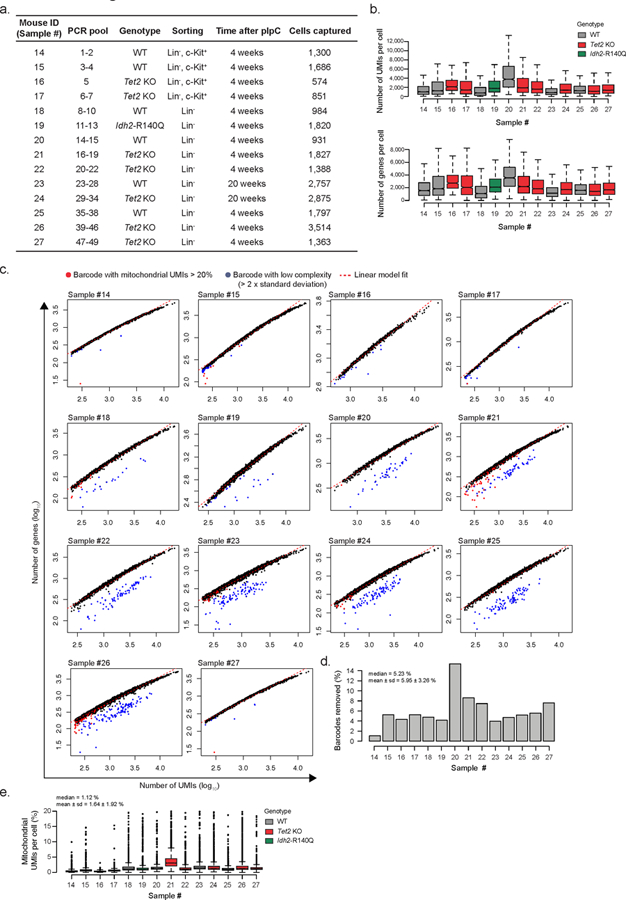
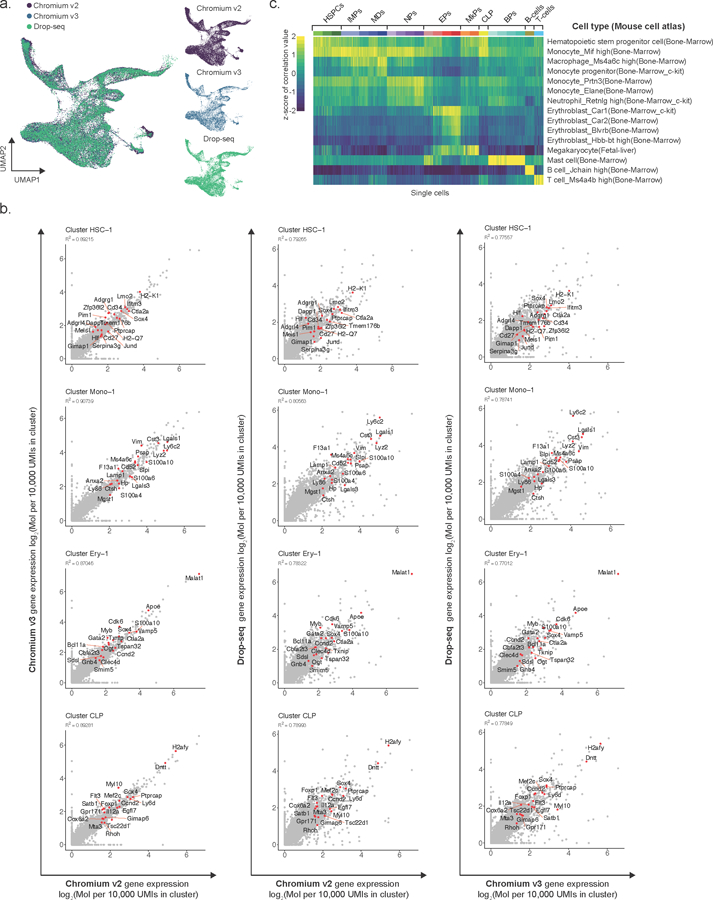
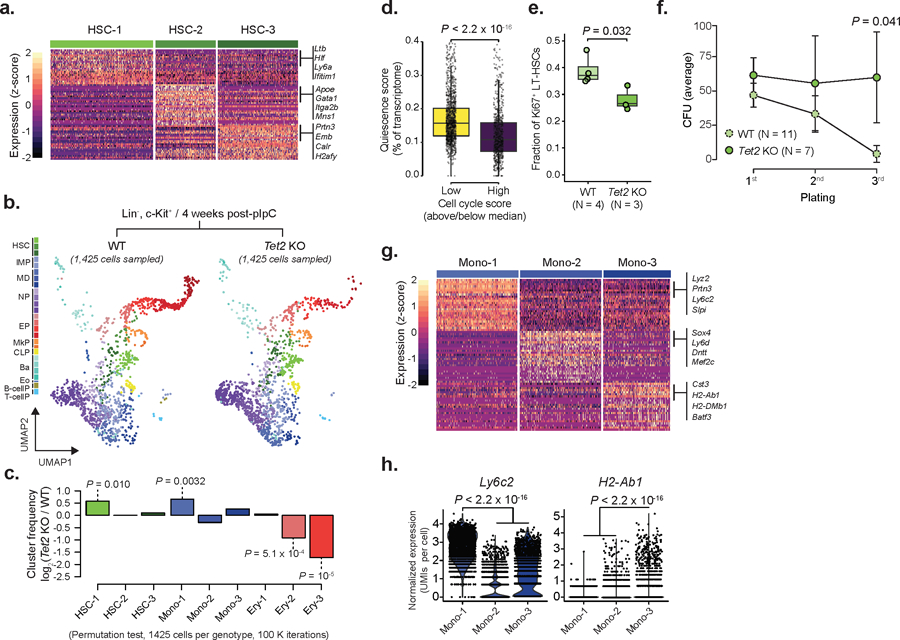
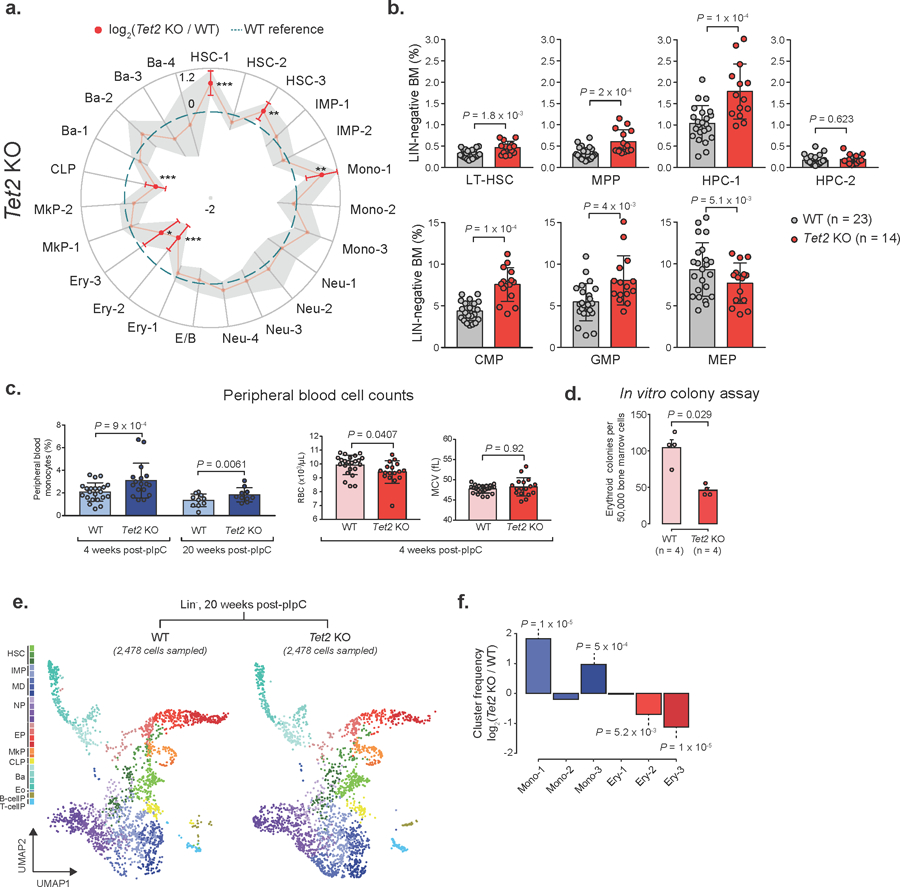


Comment in
-
DNMT3A and TET2 mutations reshape hematopoiesis in opposing ways.Nat Genet. 2020 Jun;52(6):554-556. doi: 10.1038/s41588-020-0641-2. Nat Genet. 2020. PMID: 32504054 No abstract available.
References
Methods-only references
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
