

15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation
- PMID: 32205444
- PMCID: PMC7149425
- DOI: 10.1073/pnas.1920193117
15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation
Abstract
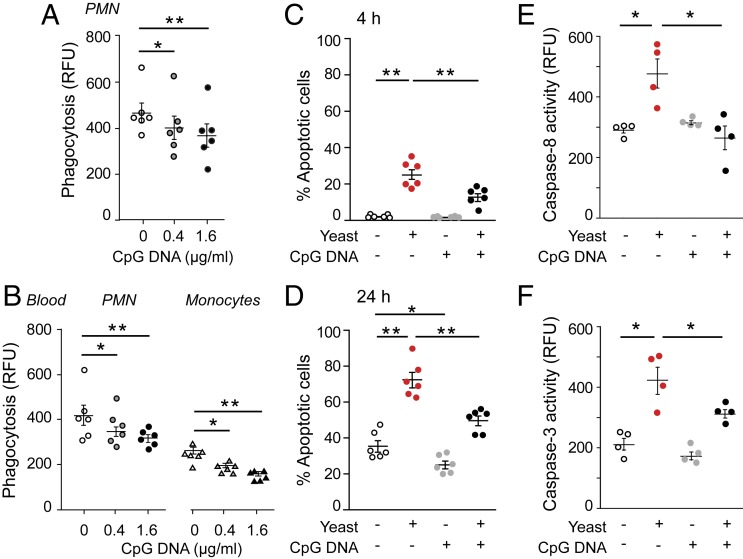
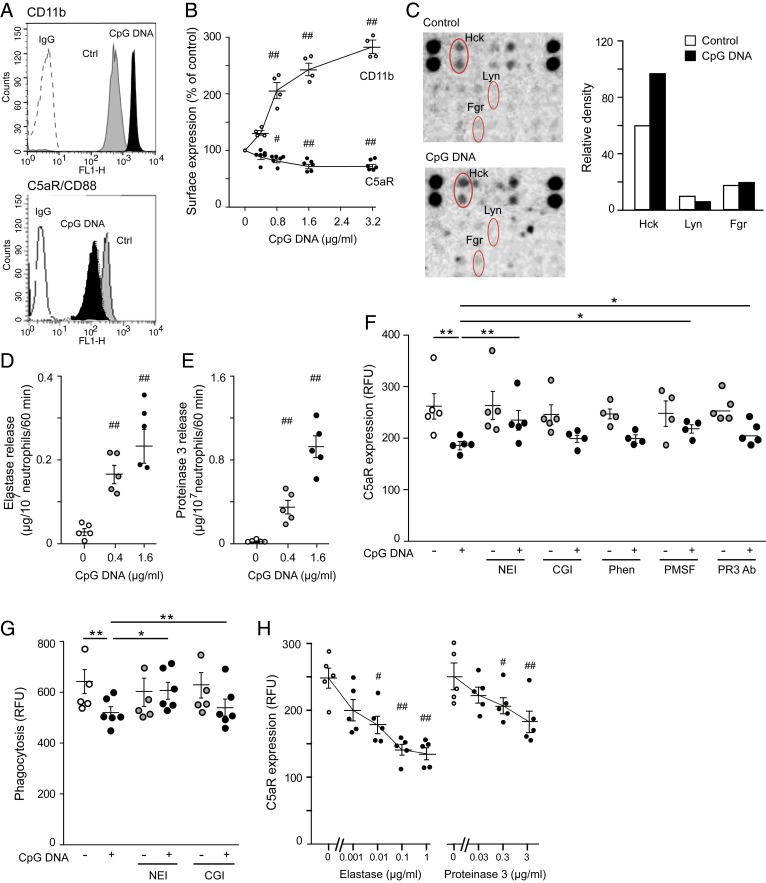
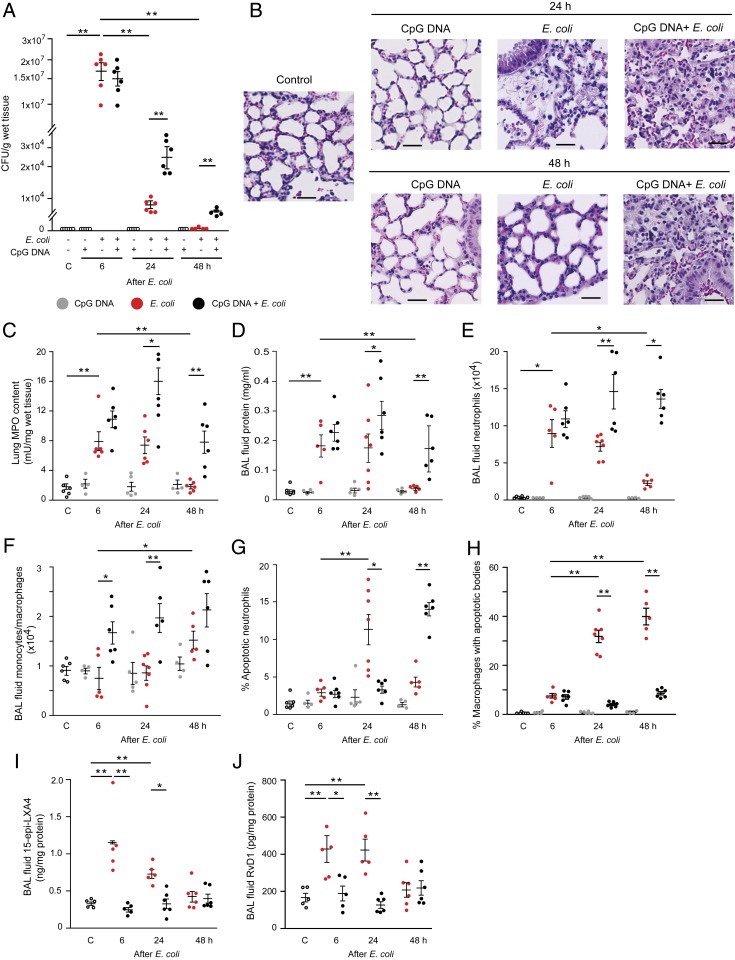
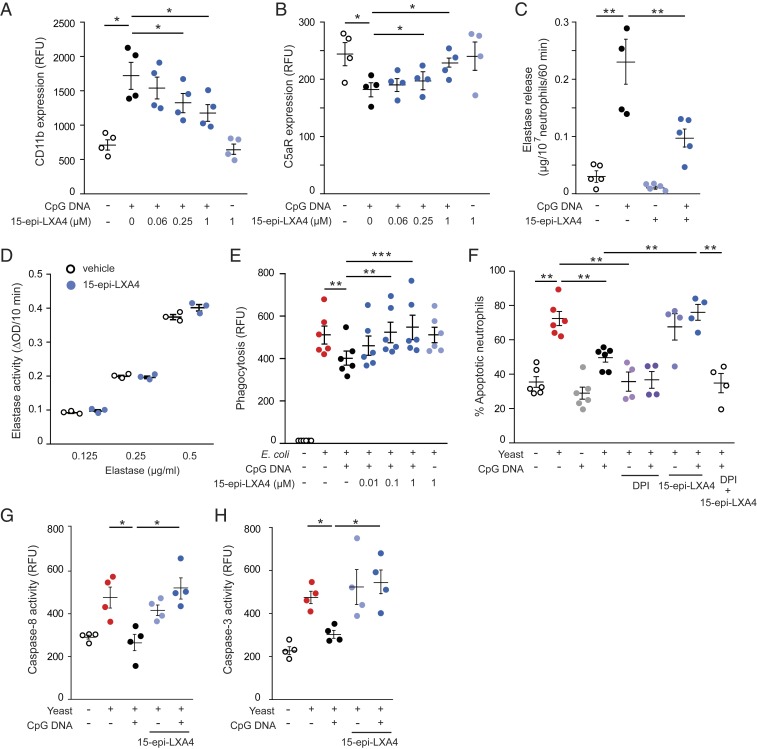
Timely resolution of bacterial infections critically depends on phagocytosis of invading pathogens by polymorphonuclear neutrophil granulocytes (PMNs), followed by PMN apoptosis and efferocytosis. Here we report that bacterial DNA (CpG DNA) and mitochondrial DNA impair phagocytosis and attenuate phagocytosis-induced apoptosis in human PMNs through Toll-like receptor 9 (TLR9)-mediated release of neutrophil elastase and proteinase 3 and subsequent down-regulation of the complement receptor C5aR. Consistently, CpG DNA delays pulmonary clearance of Escherichia coli in mice and suppresses PMN apoptosis, efferocytosis, and generation of proresolving lipid mediators, thereby prolonging lung inflammation evoked by E. coli Genetic deletion of TLR9 renders mice unresponsive to CpG DNA. We also show that aspirin-triggered 15-epi-lipoxin A4 (15-epi-LXA4) and 17-epi-resolvin D1 (17-epi-RvD1) through the receptor ALX/FPR2 antagonize cues from CpG DNA, preserve C5aR expression, restore impaired phagocytosis, and redirect human PMNs to apoptosis. Treatment of mice with 15-epi-LXA4 or 17-epi-RvD1 at the peak of inflammation accelerates clearance of bacteria, blunts PMN accumulation, and promotes PMN apoptosis and efferocytosis, thereby facilitating resolution of E. coli-evoked lung injury. Collectively, these results uncover a TLR9-mediated endogenous mechanism that impairs PMN phagocytosis and prolongs inflammation, and demonstrate both endogenous and therapeutic potential for 15-epi-LXA4 and 17-epi-RvD1 to restore impaired bacterial clearance and facilitate resolution of acute lung inflammation.
Keywords: TLR9; aspirin-triggered LXA4 and RvD1; neutrophils; phagocytosis; resolution of inflammation.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
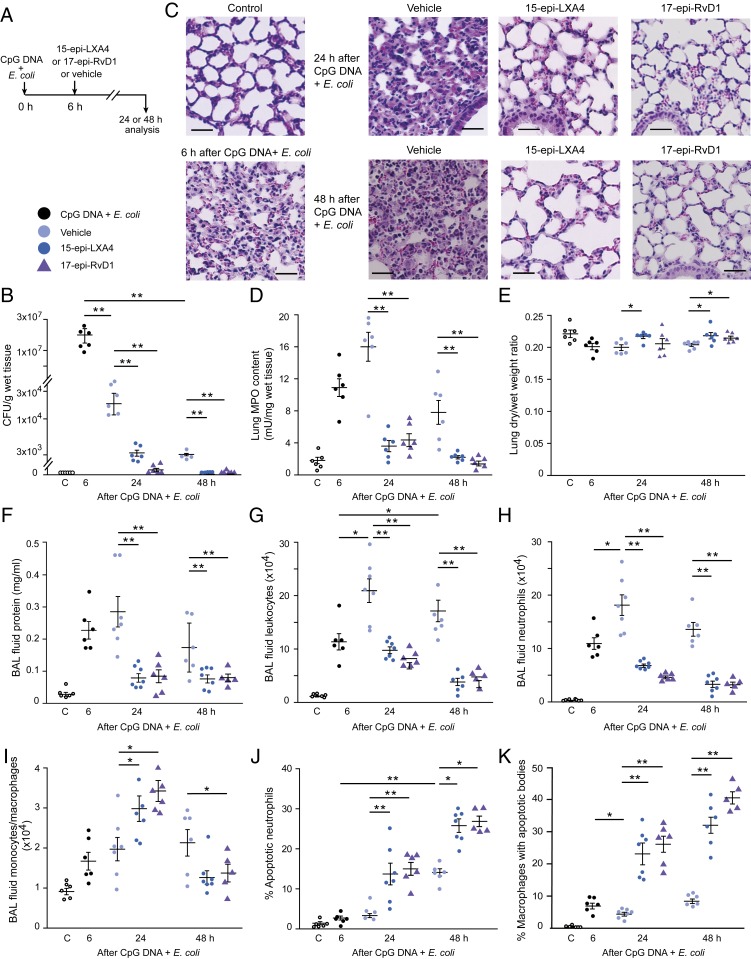
Comment in
-
Proresolving lipid mediators enhance PMN-mediated bacterial clearance.Proc Natl Acad Sci U S A. 2020 Apr 28;117(17):9148-9150. doi: 10.1073/pnas.2004241117. Epub 2020 Apr 16. Proc Natl Acad Sci U S A. 2020. PMID: 32300015 Free PMC article. No abstract available.
References
-
- Nathan C., Ding A., Nonresolving inflammation. Cell 140, 871–882 (2010). - PubMed
-
- Gilroy D. W., Lawrence T., Perretti M., Rossi A. G., Inflammatory resolution: New opportunities for drug discovery. Nat. Rev. Drug Discov. 3, 401–416 (2004). - PubMed
-
- Rossi A. G., et al. , Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064 (2006). - PubMed
-
- Hotchkiss R. S., Karl I. E., The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
