

Gene editing and CRISPR in the clinic: current and future perspectives
- PMID: 32207531
- PMCID: PMC7146048
- DOI: 10.1042/BSR20200127
Gene editing and CRISPR in the clinic: current and future perspectives
Abstract
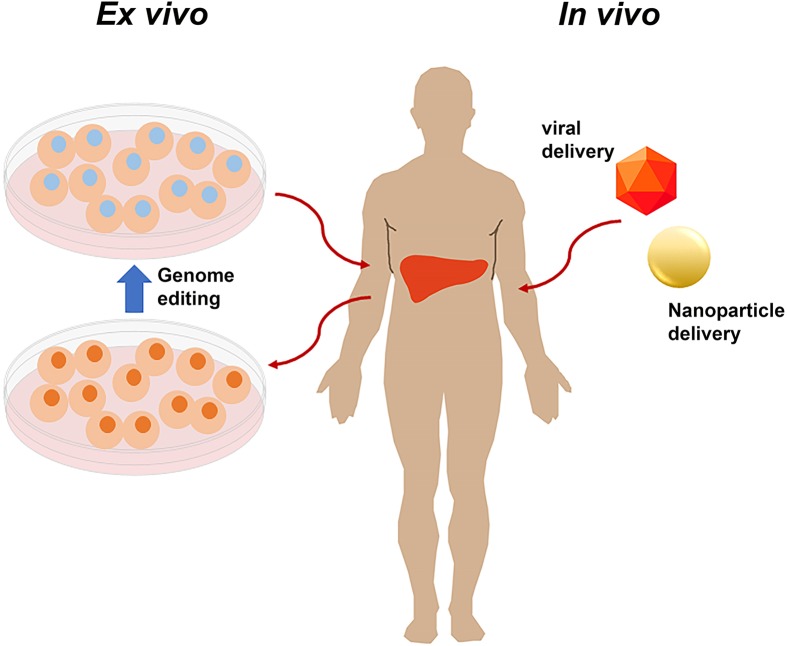
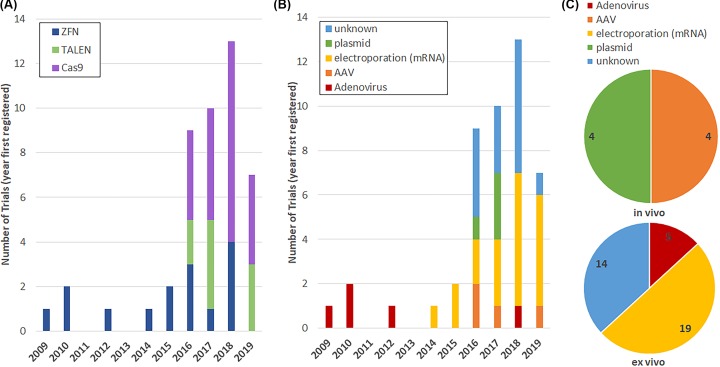
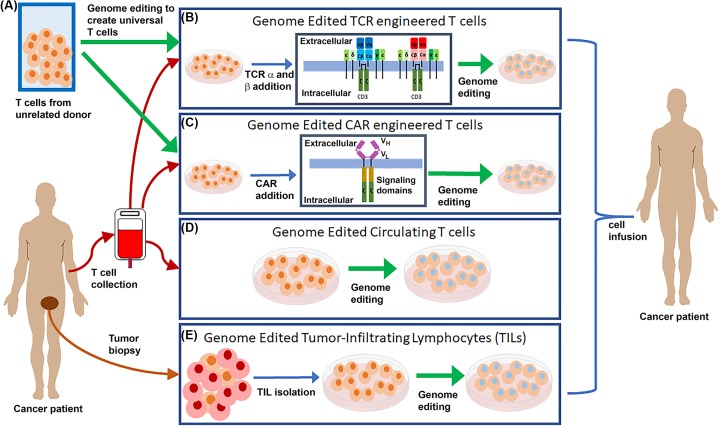
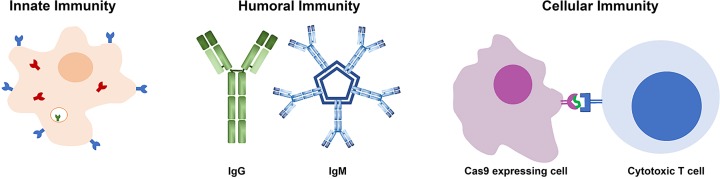
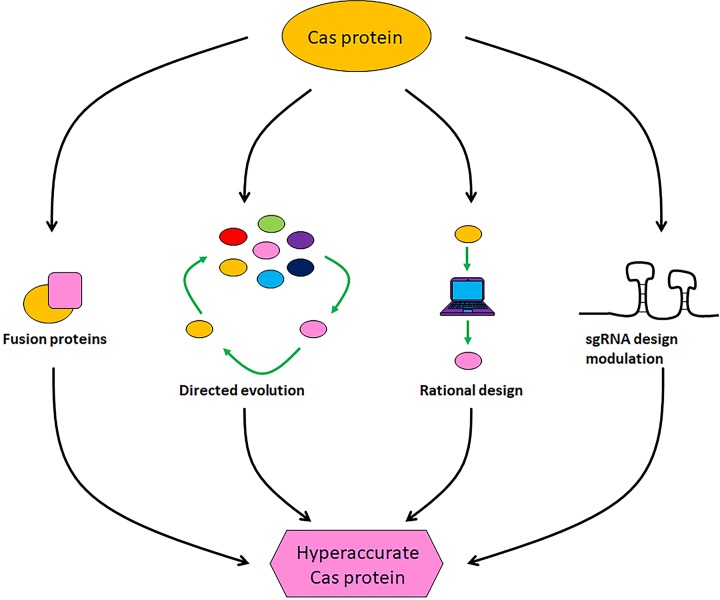
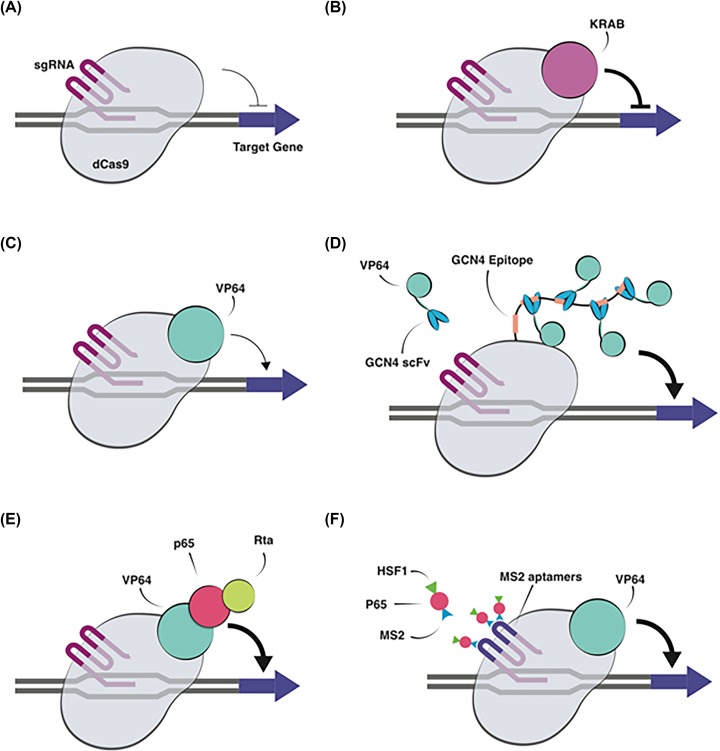
Genome editing technologies, particularly those based on zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR (clustered regularly interspaced short palindromic repeat DNA sequences)/Cas9 are rapidly progressing into clinical trials. Most clinical use of CRISPR to date has focused on ex vivo gene editing of cells followed by their re-introduction back into the patient. The ex vivo editing approach is highly effective for many disease states, including cancers and sickle cell disease, but ideally genome editing would also be applied to diseases which require cell modification in vivo. However, in vivo use of CRISPR technologies can be confounded by problems such as off-target editing, inefficient or off-target delivery, and stimulation of counterproductive immune responses. Current research addressing these issues may provide new opportunities for use of CRISPR in the clinical space. In this review, we examine the current status and scientific basis of clinical trials featuring ZFNs, TALENs, and CRISPR-based genome editing, the known limitations of CRISPR use in humans, and the rapidly developing CRISPR engineering space that should lay the groundwork for further translation to clinical application.
Keywords: CRISPR; clinical trial; gene activation; genome editing; transcription activator-like effector nucleases; zinc finger nuclease.
© 2020 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
References
-
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J. et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical