

Localized Hypermutation is the Major Driver of Meningococcal Genetic Variability during Persistent Asymptomatic Carriage
- PMID: 32209693
- PMCID: PMC7157529
- DOI: 10.1128/mBio.03068-19
Localized Hypermutation is the Major Driver of Meningococcal Genetic Variability during Persistent Asymptomatic Carriage
Abstract
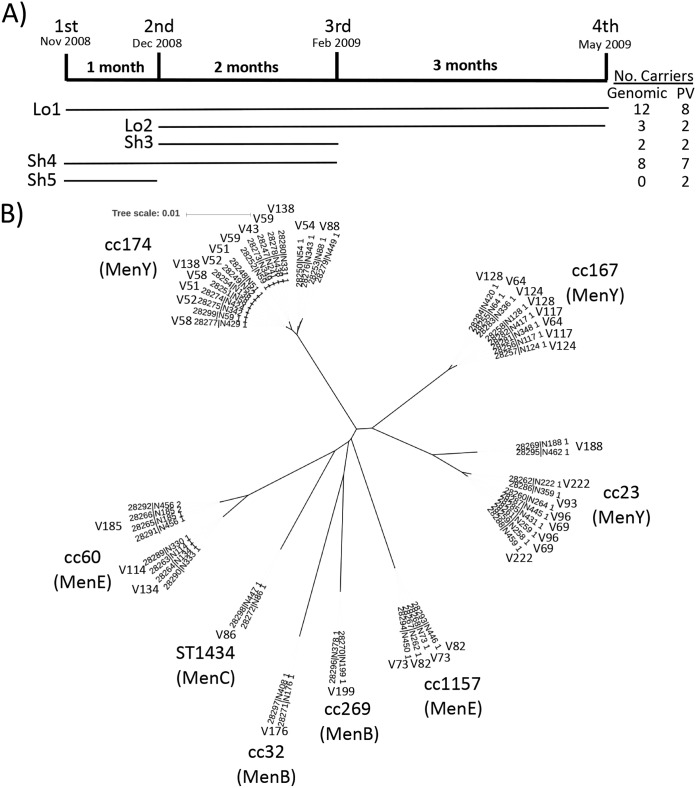
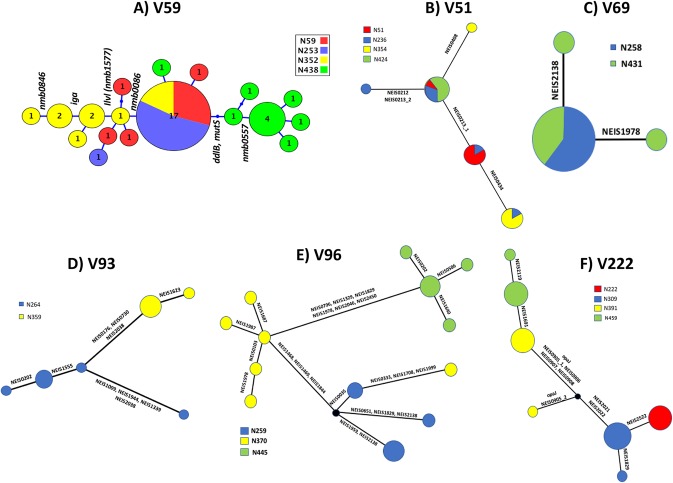
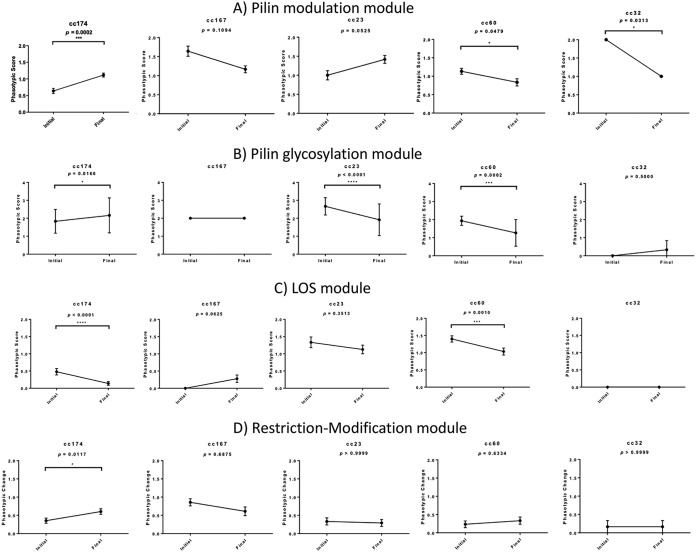
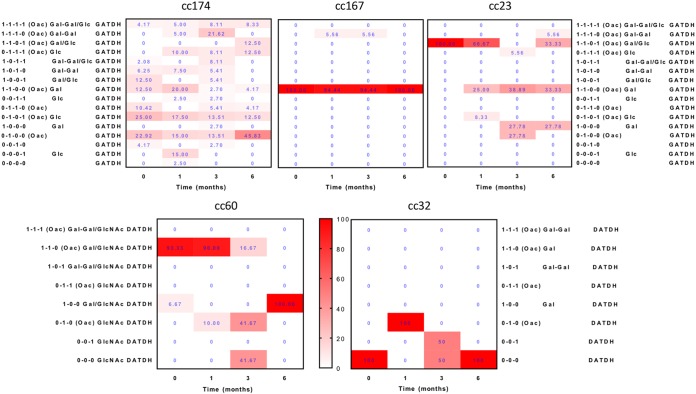
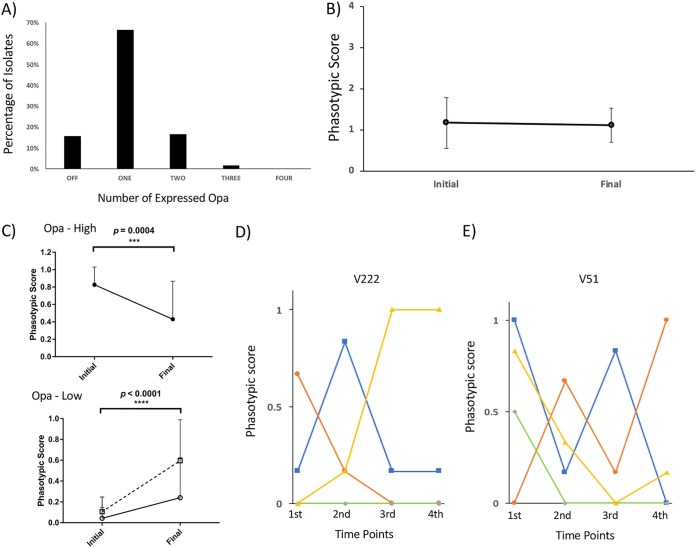
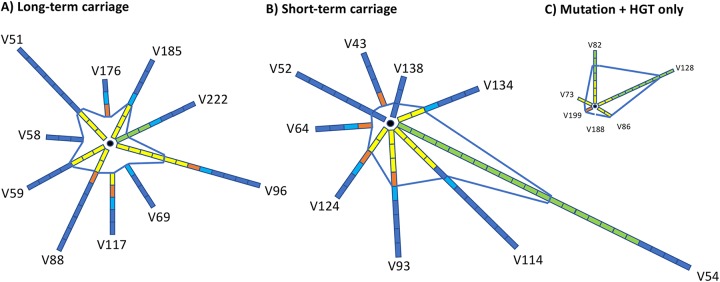
Host persistence of bacteria is facilitated by mutational and recombinatorial processes that counteract loss of genetic variation during transmission and selection from evolving host responses. Genetic variation was investigated during persistent asymptomatic carriage of Neisseria meningitidis Interrogation of whole-genome sequences for paired isolates from 25 carriers showed that de novo mutations were infrequent, while horizontal gene transfer occurred in 16% of carriers. Examination of multiple isolates per time point enabled separation of sporadic and transient allelic variation from directional variation. A comprehensive comparative analysis of directional allelic variation with hypermutation of simple sequence repeats and hyperrecombination of class 1 type IV pilus genes detected an average of seven events per carrier and 2:1 bias for changes due to localized hypermutation. Directional genetic variation was focused on the outer membrane with 69% of events occurring in genes encoding enzymatic modifiers of surface structures or outer membrane proteins. Multiple carriers exhibited directional and opposed switching of allelic variants of the surface-located Opa proteins that enables continuous expression of these adhesins alongside antigenic variation. A trend for switching from PilC1 to PilC2 expression was detected, indicating selection for specific alterations in the activities of the type IV pilus, whereas phase variation of restriction modification (RM) systems, as well as associated phasevarions, was infrequent. We conclude that asymptomatic meningococcal carriage on mucosal surfaces is facilitated by frequent localized hypermutation and horizontal gene transfer affecting genes encoding surface modifiers such that optimization of adhesive functions occurs alongside escape of immune responses by antigenic variation.IMPORTANCE Many bacterial pathogens coexist with host organisms, rarely causing disease while adapting to host responses. Neisseria meningitidis, a major cause of meningitis and septicemia, is a frequent persistent colonizer of asymptomatic teenagers/young adults. To assess how genetic variation contributes to host persistence, whole-genome sequencing and hypermutable sequence analyses were performed on multiple isolates obtained from students naturally colonized with meningococci. High frequencies of gene transfer were observed, occurring in 16% of carriers and affecting 51% of all nonhypermutable variable genes. Comparative analyses showed that hypermutable sequences were the major mechanism of variation, causing 2-fold more changes in gene function than other mechanisms. Genetic variation was focused on genes affecting the outer membrane, with directional changes in proteins responsible for bacterial adhesion to host surfaces. This comprehensive examination of genetic plasticity in individual hosts provides a significant new platform for rationale design of approaches to prevent the spread of this pathogen.
Keywords: Neisseria meningitidis; horizontal gene transfer; localized hypermutation; meningitis; meningococcus; phase variation; whole-genome sequence.
Copyright © 2020 Green et al.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
