

A protein interaction free energy model based on amino acid residue contributions: Assessment of point mutation stability of T4 lysozyme
- PMID: 32211456
- PMCID: PMC7093156
- DOI: 10.1142/s233954781950002x
A protein interaction free energy model based on amino acid residue contributions: Assessment of point mutation stability of T4 lysozyme
Abstract
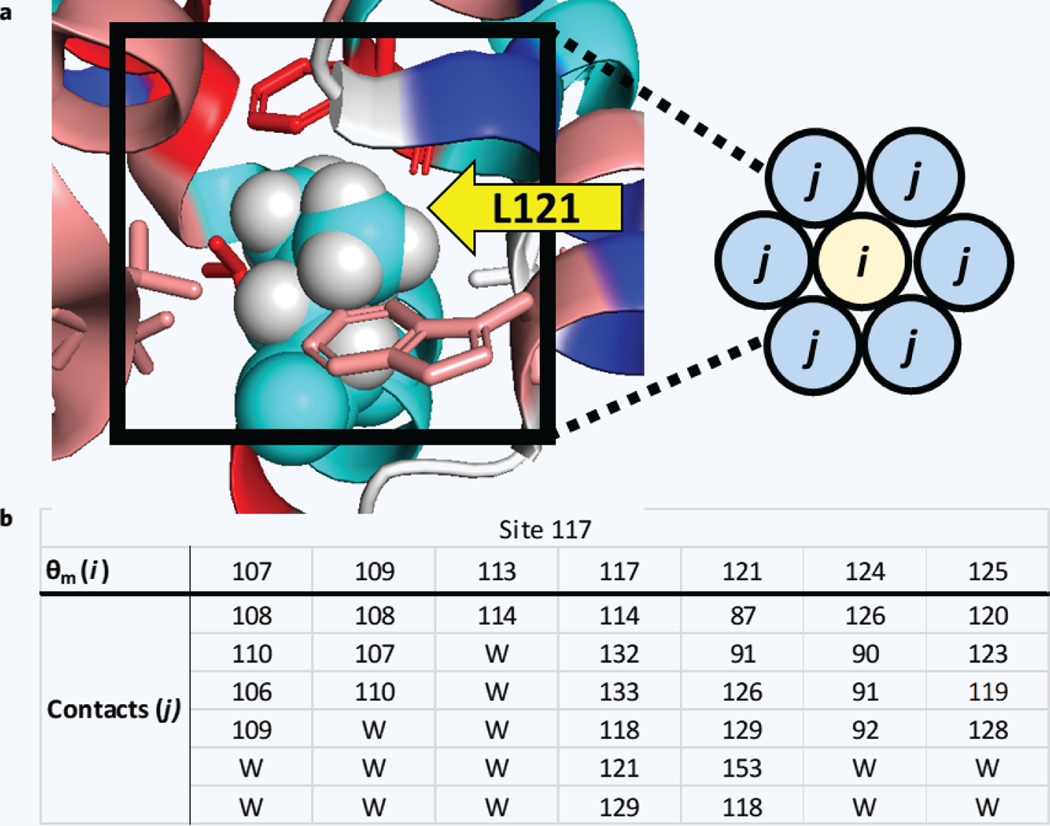
Here we present a model to estimate the interaction free energy contribution of each amino acid residue of a given protein. Protein interaction energy is described in terms of per-residue interaction factors, μ. Multibody interactions are implicitly captured in μ through the combination of amino acid terms (γ) guided by local conformation indices (σ). The model enables construction of an interaction factor heat map for a protein in a given fold, allows prima facie assessment of the degree of residue-residue interaction, and facilitates a qualitative and quantitative evaluation of protein association properties. The model was used to compute thermal stability of T4 bacteriophage lysozyme mutants across seven sites. Qualitative assessment of mutational effects provides a straightforward rationale regarding whether a particular site primarily perturbs native or non-native states, or both. The presented model was found to be in good agreement with experimental mutational data (R 2 = 0.73) and suggests an approach by which to convert structure space into energy space.
Keywords: Coarse-Grain Protein Model; Peptide Engineering; Protein Engineering; Protein Interaction Free Energy; Protein Thermal Stability.
Figures






References
-
- Carlson R Estimating the biotech sector’s contribution to the US economy. Nature Biotechnol. 34, 247 (2016). - PubMed
-
- Burley SK et al. In Protein Crystallography: Methods and Protocols. (eds.) Wlodawer A, Dauter Z & Jaskolski M. (Springer, New York, pp. 627–641, 2017).
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials