

Heterozygous STUB1 missense variants cause ataxia, cognitive decline, and STUB1 mislocalization
- PMID: 32211513
- PMCID: PMC7073456
- DOI: 10.1212/NXG.0000000000000397
Heterozygous STUB1 missense variants cause ataxia, cognitive decline, and STUB1 mislocalization
Abstract
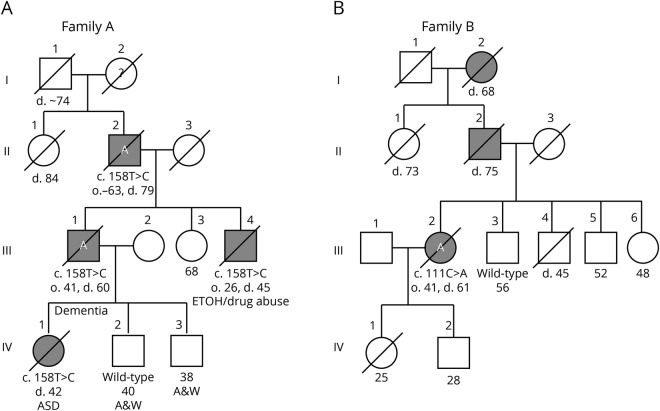
Objective: To identify the genetic cause of autosomal dominant ataxia complicated by behavioral abnormalities, cognitive decline, and autism in 2 families and to characterize brain neuropathologic signatures of dominant STUB1-related ataxia and investigate the effects of pathogenic variants on STUB1 localization.
Methods: Clinical and research-based exome sequencing was used to identify the causative variants for autosomal dominant ataxia in 2 families. Gross and microscopic neuropathologic evaluations were performed on the brains of 4 affected individuals in these families.
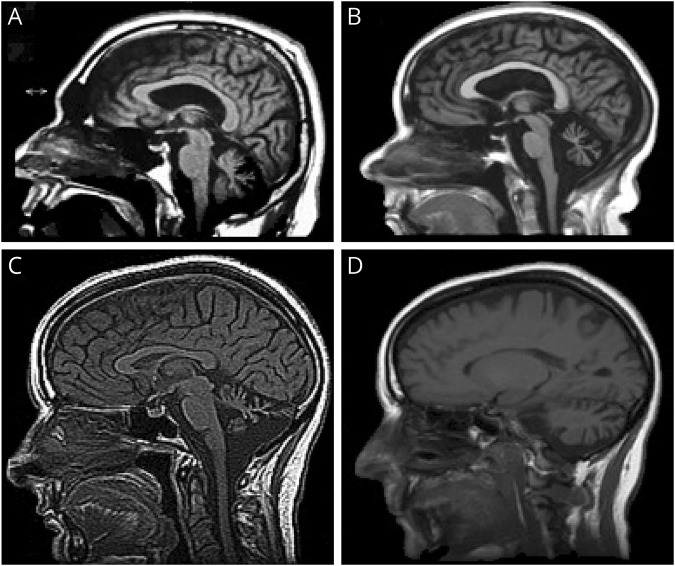
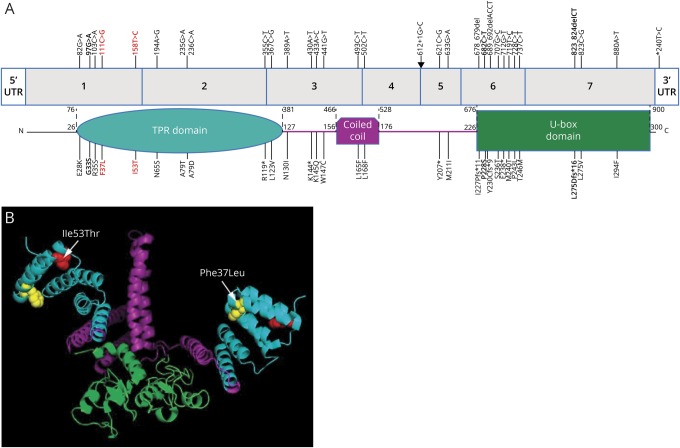
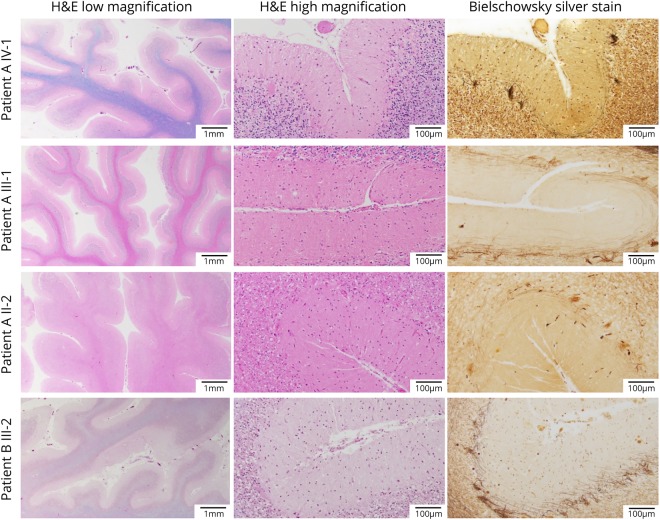
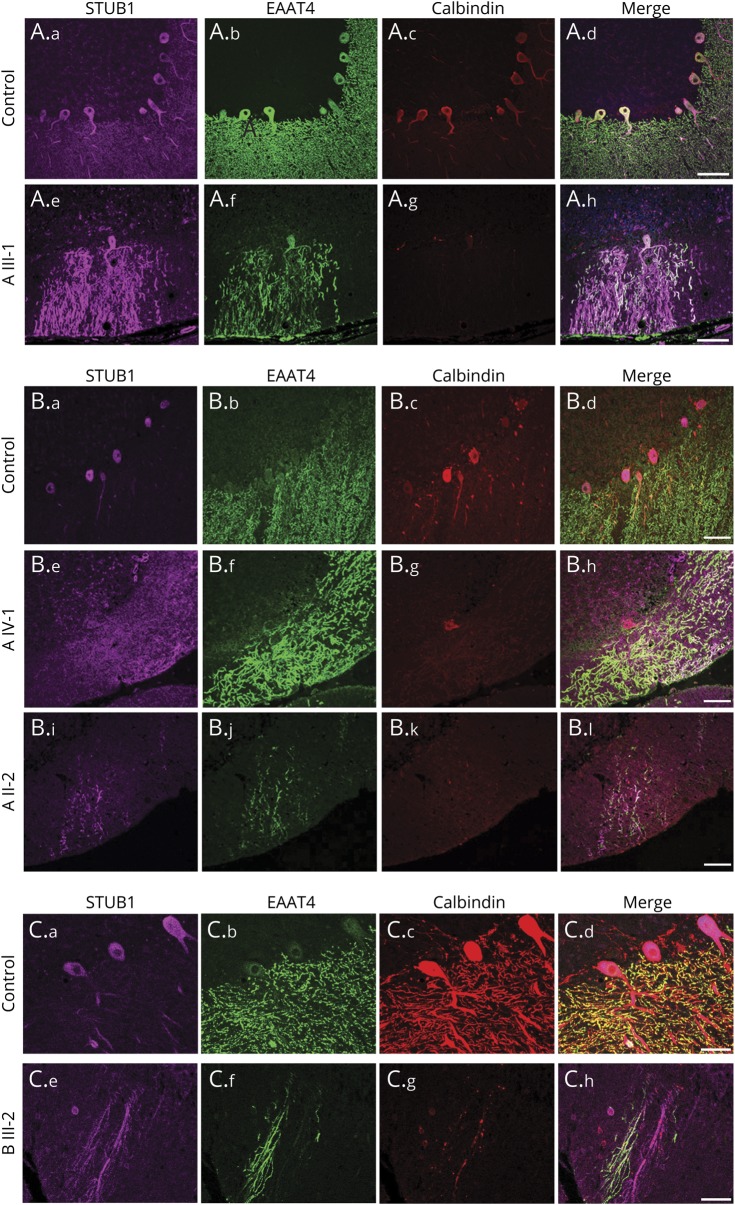
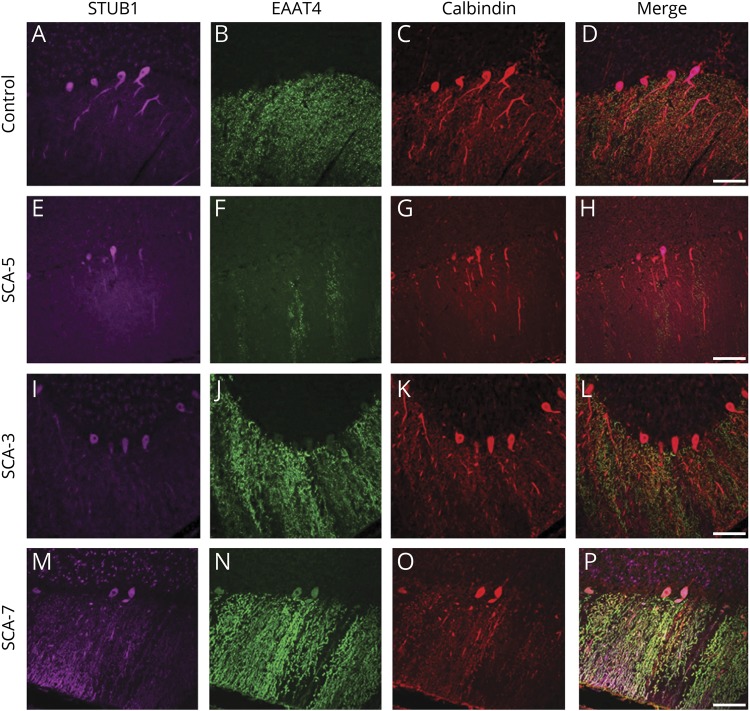
Results: Mutations in STUB1 have been primarily associated with childhood-onset autosomal recessive ataxia, but here we report heterozygous missense variants in STUB1 (p.Ile53Thr and p.The37Leu) confirming the recent reports of autosomal dominant inheritance. Cerebellar atrophy on imaging and cognitive deficits often preceded ataxia. Unique neuropathologic examination of the 4 brains showed the marked loss of Purkinje cells (PCs) without microscopic evidence of significant pathology outside the cerebellum. The normal pattern of polarized somatodendritic STUB1 protein expression in PCs was lost, resulting in aberrant STUB1 localization in the distal PC dendritic arbors.
Conclusions: This study confirms a dominant inheritance pattern in STUB1-ataxia in addition to a recessive one and documents its association with cognitive and behavioral disability, including autism. In the most extensive analysis of cerebellar pathology in this disease, we demonstrate disruption of STUB1 protein in PCs as part of the underlying pathogenesis.
Copyright © 2020 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
References
-
- Bird T. Hereditary ataxia overview. In: GeneReviews at GeneTests: Medical Genetics Information Resource [Database Online] 1993-2019. Seattle: University of Washington; 2019.
-
- Depondt C, Donatello S, Simonis N, et al. . Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology 2014;82:1749–1750. - PubMed
-
- Bettencourt C, de Yebenes JG, Lopez-Sendon JL, et al. . Clinical and neuropathological features of spastic ataxia in a Spanish family with novel compound heterozygous mutations in STUB1. Cerebellum 2015;14:378–381. - PubMed
-
- Genis D, Ortega-Cubero S, San Nicolas H, et al. . Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 2018;91:e1988–e1998. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Medical