

How We Think about Targeting RNA with Small Molecules
- PMID: 32212706
- PMCID: PMC7486258
- DOI: 10.1021/acs.jmedchem.9b01927
How We Think about Targeting RNA with Small Molecules
Abstract
RNA offers nearly unlimited potential as a target for small molecule chemical probes and lead medicines. Many RNAs fold into structures that can be selectively targeted with small molecules. This Perspective discusses molecular recognition of RNA by small molecules and highlights key enabling technologies and properties of bioactive interactions. Sequence-based design of ligands targeting RNA has established rules for affecting RNA targets and provided a potentially general platform for the discovery of bioactive small molecules. The RNA targets that contain preferred small molecule binding sites can be identified from sequence, allowing identification of off-targets and prediction of bioactive interactions by nature of ligand recognition of functional sites. Small molecule targeted degradation of RNA targets (ribonuclease-targeted chimeras, RIBOTACs) and direct cleavage by small molecules have also been developed. These growing technologies suggest that the time is right to provide small molecule chemical probes to target functionally relevant RNAs throughout the human transcriptome.
Conflict of interest statement
The authors declare the following competing financial interest(s): M.D.D. is a founder of Expansion Therapeutics.
Figures

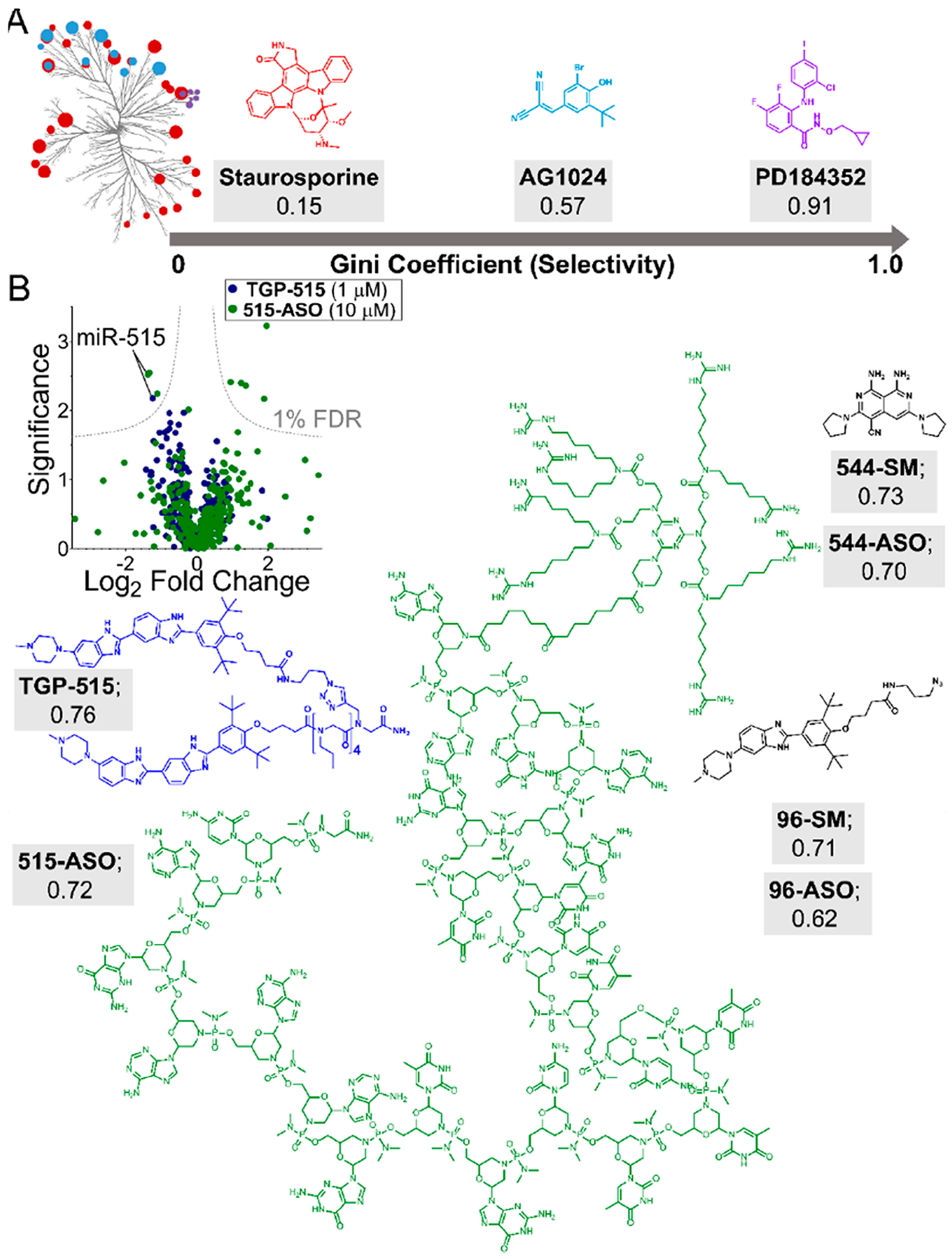
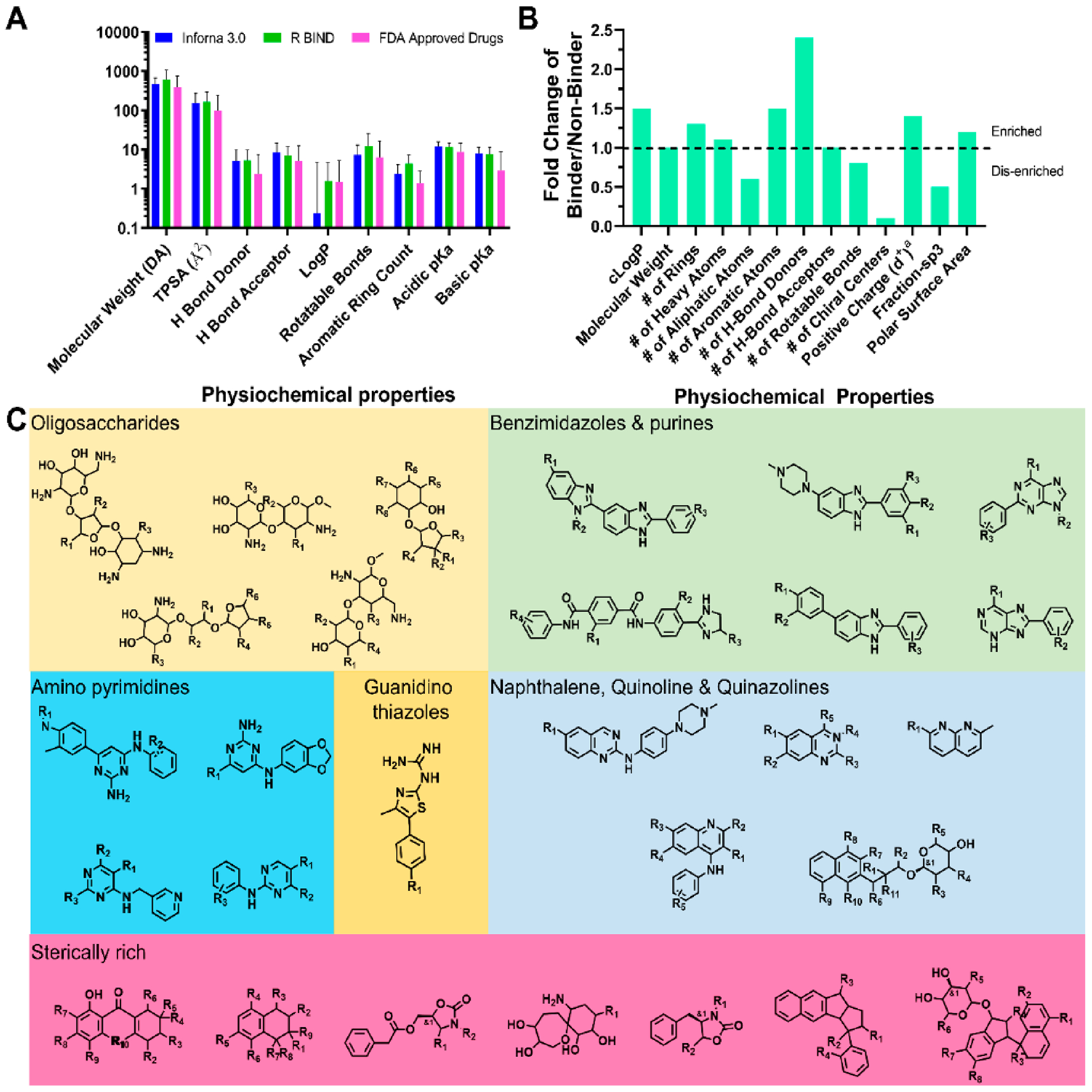
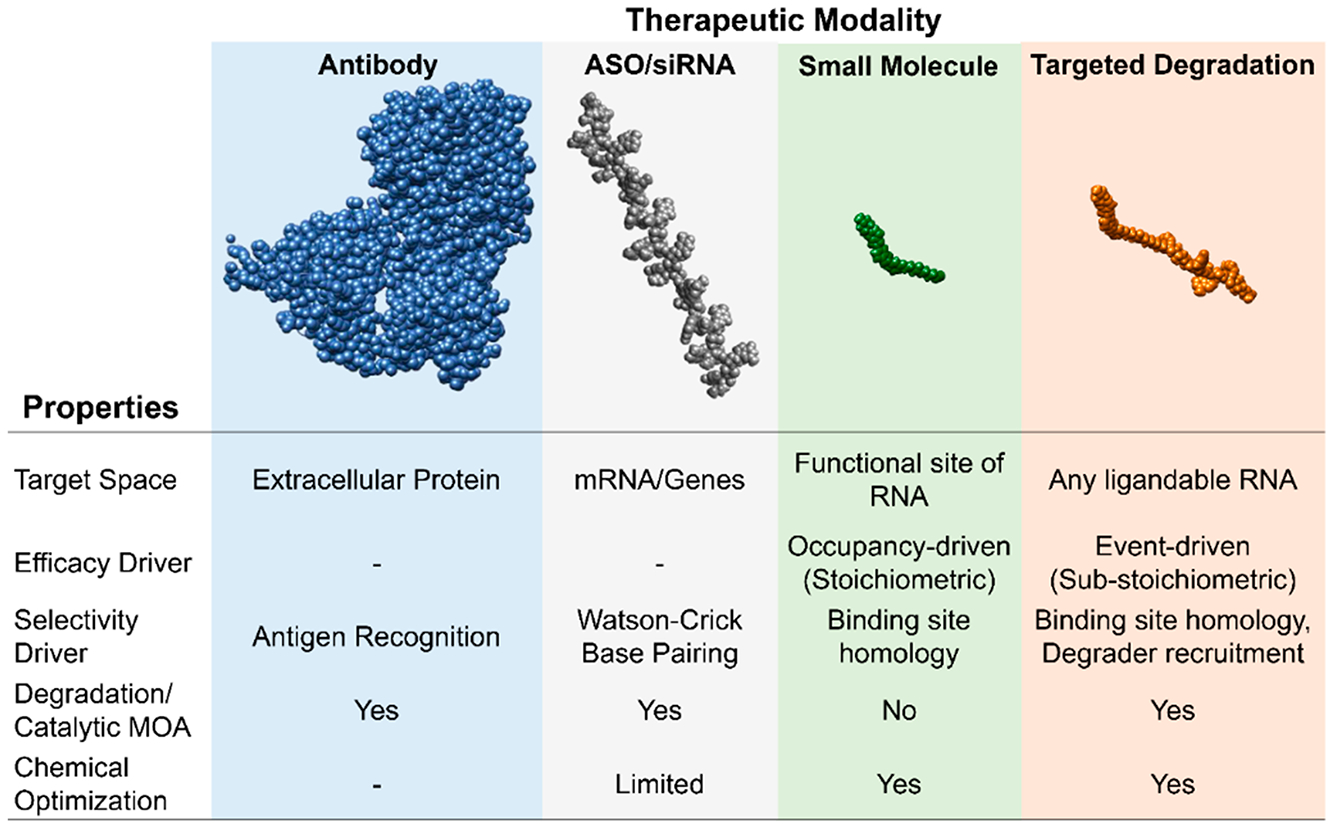
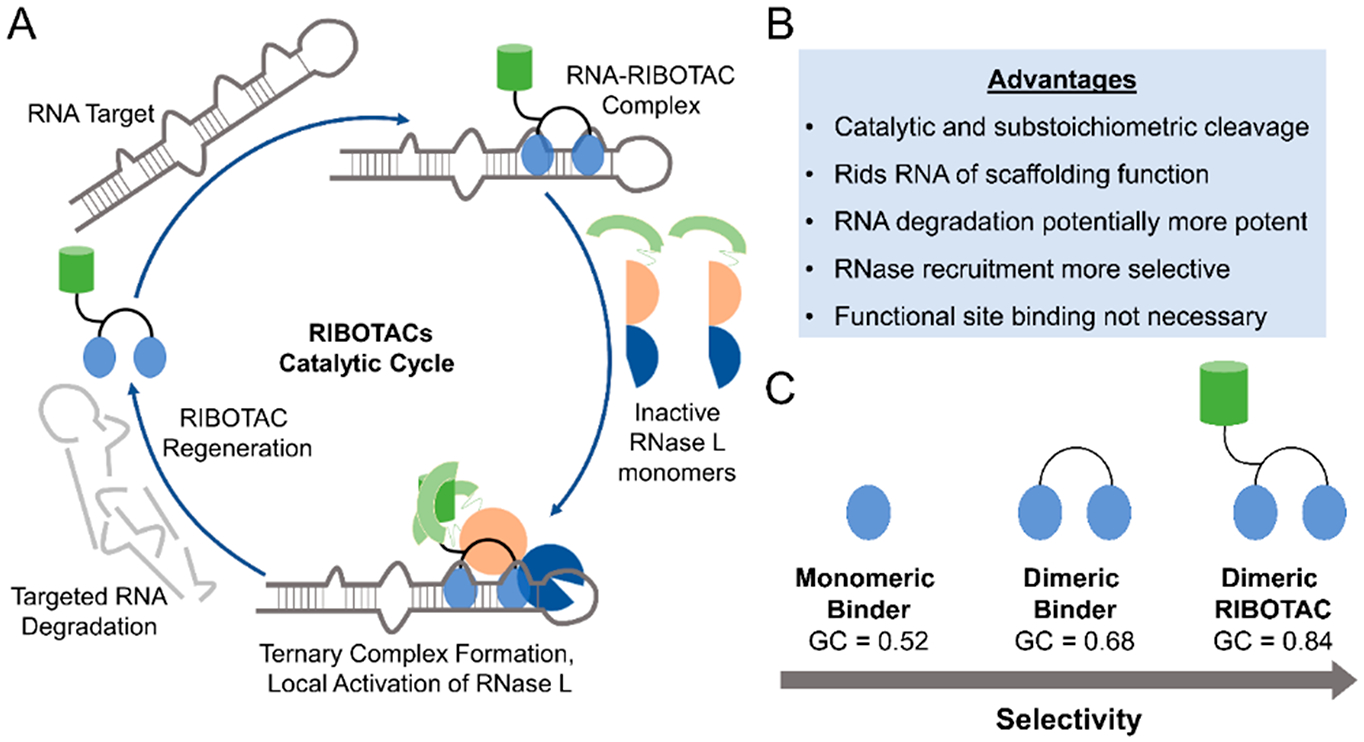
References
-
- Lander ES; Linton LM; Birren B; Nusbaum C; Zody MC; Baldwin J; Devon K; Dewar K; Doyle M; FitzHugh W; Funke R; Gage D; Harris K; Heaford A; Howland J; Kann L; Lehoczky J; LeVine R; McEwan P; McKernan K; Meldrim J; Mesirov JP; Miranda C; Morris W; Naylor J; Raymond C; Rosetti M; Santos R; Sheridan A; Sougnez C; Stange-Thomann Y; Stojanovic N; Subramanian A; Wyman D; Rogers J; Sulston J; Ainscough R; Beck S; Bentley D; Burton J; Clee C; Carter N; Coulson A; Deadman R; Deloukas P; Dunham A; Dunham I; Durbin R; French L; Grafham D; Gregory S; Hubbard T; Humphray S; Hunt A; Jones M; Lloyd C; McMurray A; Matthews L; Mercer S; Milne S; Mullikin JC; Mungall A; Plumb R; Ross M; Shownkeen R; Sims S; Waterston RH; Wilson RK; Hillier LW; McPherson JD; Marra MA; Mardis ER; Fulton LA; Chinwalla AT; Pepin KH; Gish WR; Chissoe SL; Wendl MC; Delehaunty KD; Miner TL; Delehaunty A; Kramer JB; Cook LL; Fulton RS; Johnson DL; Minx PJ; Clifton SW; Hawkins T; Branscomb E; Predki P; Richardson P; Wenning S; Slezak T; Doggett N; Cheng JF; Olsen A; Lucas S; Elkin C; Uberbacher E; Frazier M; Gibbs RA; Muzny DM; Scherer SE; Bouck JB; Sodergren EJ; Worley KC; Rives CM; Gorrell JH; Metzker ML; Naylor SL; Kucherlapati RS; Nelson DL; Weinstock GM; Sakaki Y; Fujiyama A; Hattori M; Yada T; Toyoda A; Itoh T; Kawagoe C; Watanabe H; Totoki Y; Taylor T; Weissenbach J; Heilig R; Saurin W; Artiguenave F; Brottier P; Bruls T; Pelletier E; Robert C; Wincker P; Smith DR; Doucette-Stamm L; Rubenfield M; Weinstock K; Lee HM; Dubois J; Rosenthal A; Platzer M; Nyakatura G; Taudien S; Rump A; Yang H; Yu J; Wang J; Huang G; Gu J; Hood L; Rowen L; Madan A; Qin S; Davis RW; Federspiel NA; Abola AP; Proctor MJ; Myers RM; Schmutz J; Dickson M; Grimwood J; Cox DR; Olson MV; Kaul R; Raymond C; Shimizu N; Kawasaki K; Minoshima S; Evans GA; Athanasiou M; Schultz R; Roe BA; Chen F; Pan H; Ramser J; Lehrach H; Reinhardt R; McCombie WR; de la Bastide M; Dedhia N; Blocker H; Hornischer K; Nordsiek G; Agarwala R; Aravind L; Bailey JA; Bateman A; Batzoglou S; Birney E; Bork P; Brown DG; Burge CB; Cerutti L; Chen HC; Church D; Clamp M; Copley RR; Doerks T; Eddy SR; Eichler EE; Furey TS; Galagan J; Gilbert JG; Harmon C; Hayashizaki Y; Haussler D; Hermjakob H; Hokamp K; Jang W; Johnson LS; Jones TA; Kasif S; Kaspryzk A; Kennedy S; Kent WJ; Kitts P; Koonin EV; Korf I; Kulp D; Lancet D; Lowe TM; McLysaght A; Mikkelsen T; Moran JV; Mulder N; Pollara VJ; Ponting CP; Schuler G; Schultz J; Slater G; Smit AF; Stupka E; Szustakowki J; Thierry-Mieg D; Thierry-Mieg J; Wagner L; Wallis J; Wheeler R; Williams A; Wolf YI; Wolfe KH; Yang SP; Yeh RF; Collins F; Guyer MS; Peterson J; Felsenfeld A; Wetterstrand KA; Patrinos A; Morgan MJ; de Jong P; Catanese JJ; Osoegawa K; Shizuya H; Choi S; Chen YJ; Szustakowki J Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. - PubMed
-
- Anderson NL; Anderson NG Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis 1998, 19, 1853–1861. - PubMed
-
- Bertozzi CR; Sasisekharan R Glycomics In Essentials of Glycobiology, 2nd ed.; Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, Eds.; Cold Spring Harbor Laboratory Press: New York, 2009. - PubMed
-
- McGettigan PA Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol 2013, 17, 4–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
