

Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy
- PMID: 32213135
- PMCID: PMC7121924
- DOI: 10.1161/CIRCRESAHA.119.314681
Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy
Abstract
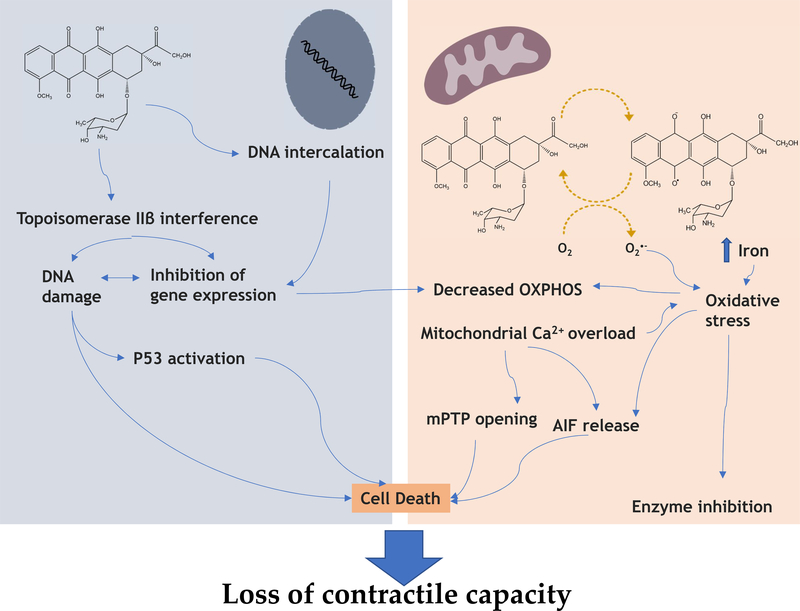
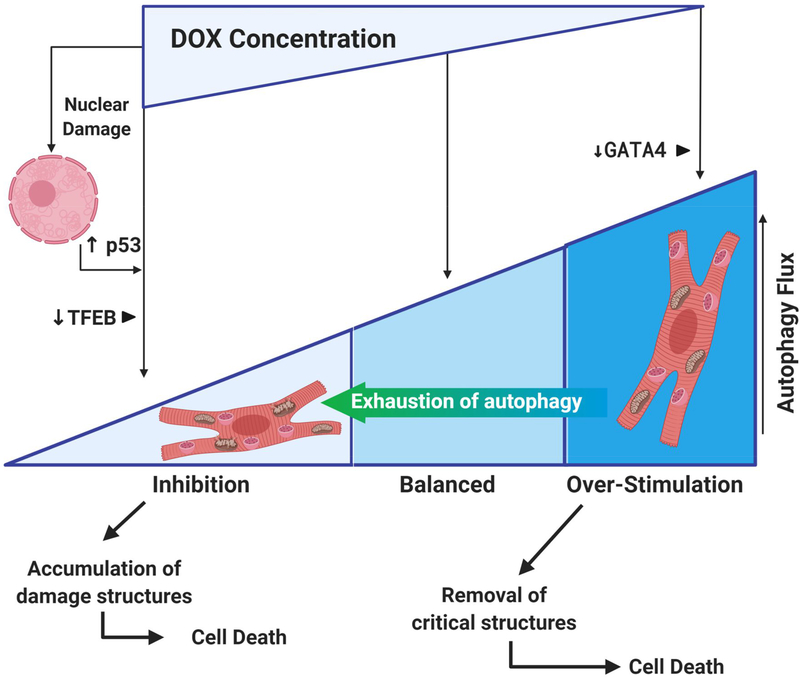
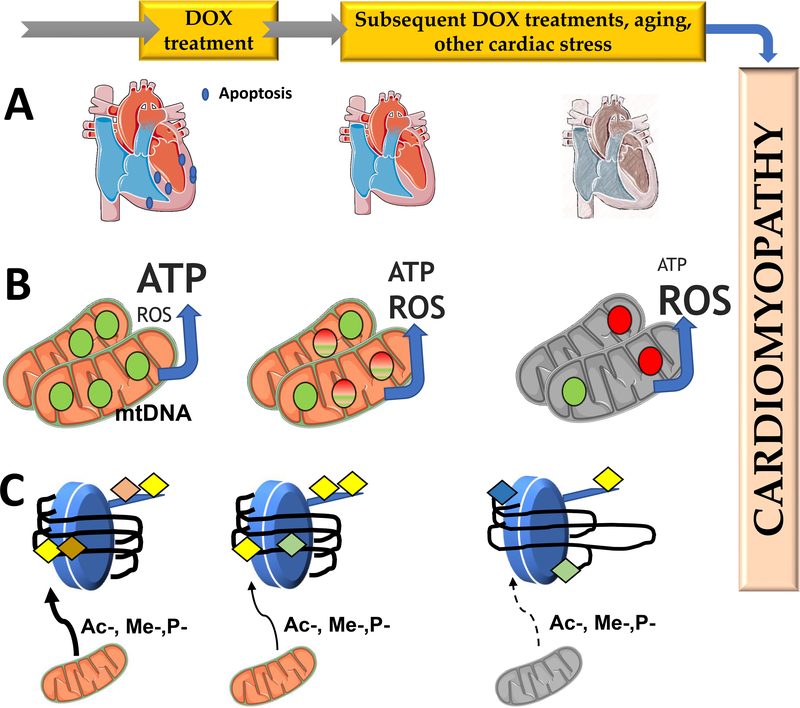
Anthracycline-based chemotherapy can result in the development of a cumulative and progressively developing cardiomyopathy. Doxorubicin is one of the most highly prescribed anthracyclines in the United States due to its broad spectrum of therapeutic efficacy. Interference with different mitochondrial processes is chief among the molecular and cellular determinants of doxorubicin cardiotoxicity, contributing to the development of cardiomyopathy. The present review provides the basis for the involvement of mitochondrial toxicity in the different functional hallmarks of anthracycline toxicity. Our objective is to understand the molecular determinants of a progressive deterioration of functional integrity of mitochondria that establishes a historic record of past drug treatments (mitochondrial memory) and renders the cancer patient susceptible to subsequent regimens of drug therapy. We focus on the involvement of doxorubicin-induced mitochondrial oxidative stress, disruption of mitochondrial oxidative phosphorylation, and permeability transition, contributing to altered metabolic and redox circuits in cardiac cells, ultimately culminating in disturbances of autophagy/mitophagy fluxes and increased apoptosis. We also suggest some possible pharmacological and nonpharmacological interventions that can reduce mitochondrial damage. Understanding the key role of mitochondria in doxorubicin-induced cardiomyopathy is essential to reduce the barriers that so dramatically limit the clinical success of this essential anticancer chemotherapy.
Keywords: cardiomyopathy; cardiotoxicity; doxorubicin; mitochondria; oxidative stress.
Figures


References
-
- Weiss RB. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol 1992;19:670–686 - PubMed
-
- Bonadonna G, Monfardini S, De Lena M, Fossati-Bellani F, Beretta G. Phase i and preliminary phase ii evaluation of adriamycin (nsc 123127). Cancer Res. 1970;30:2572–2582 - PubMed
-
- Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs 1997;54 Suppl 4:1–7 - PubMed
-
- Aubel-Sadron G, Londos-Gagliardi D. Daunorubicin and doxorubicin, anthracycline antibiotics, a physicochemical and biological review. Biochimie. 1984;66:333–352 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
