

Ultrastructural Details of Mammalian Chromosome Architecture
- PMID: 32213324
- PMCID: PMC7222625
- DOI: 10.1016/j.molcel.2020.03.003
Ultrastructural Details of Mammalian Chromosome Architecture
Abstract
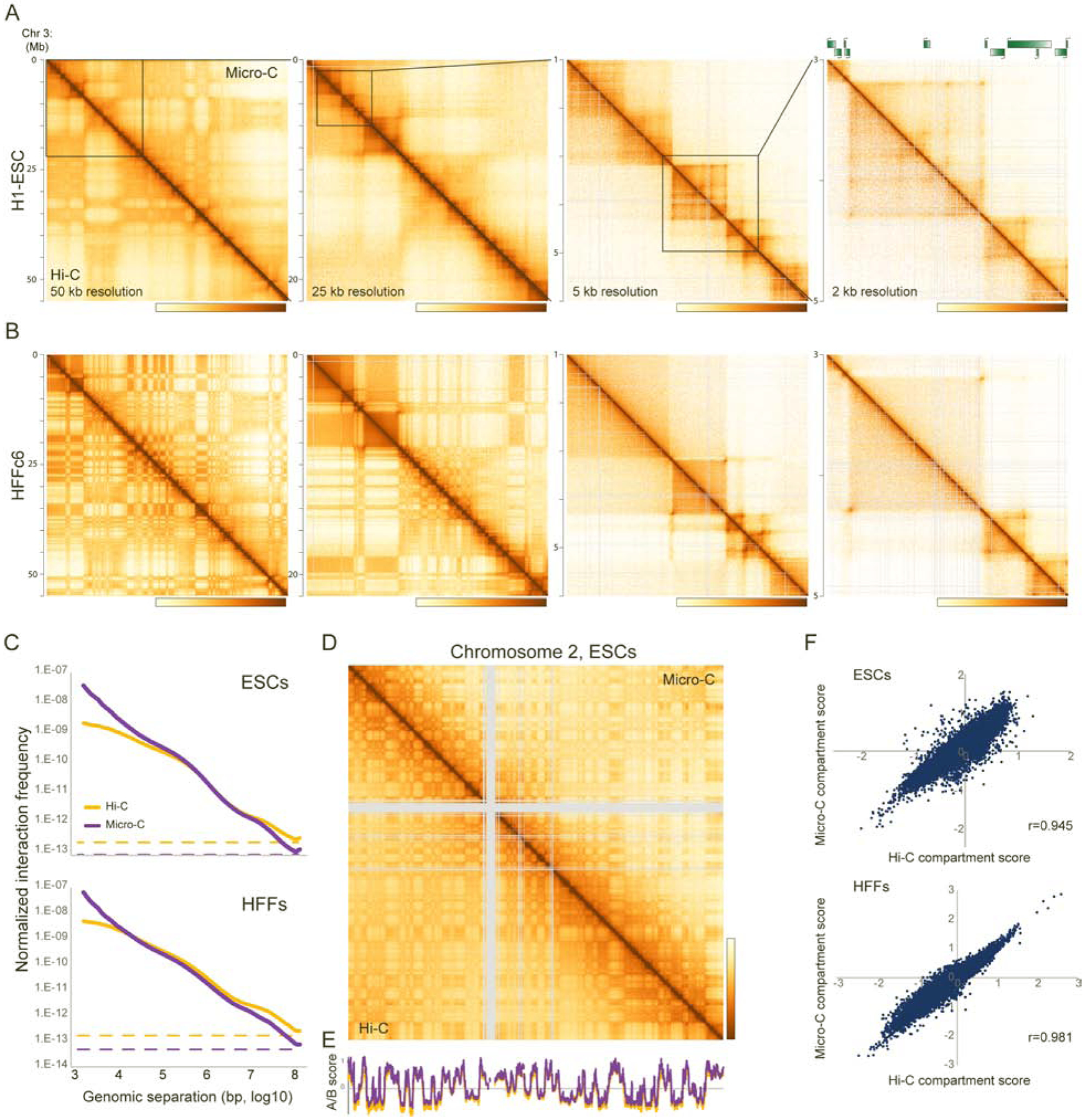
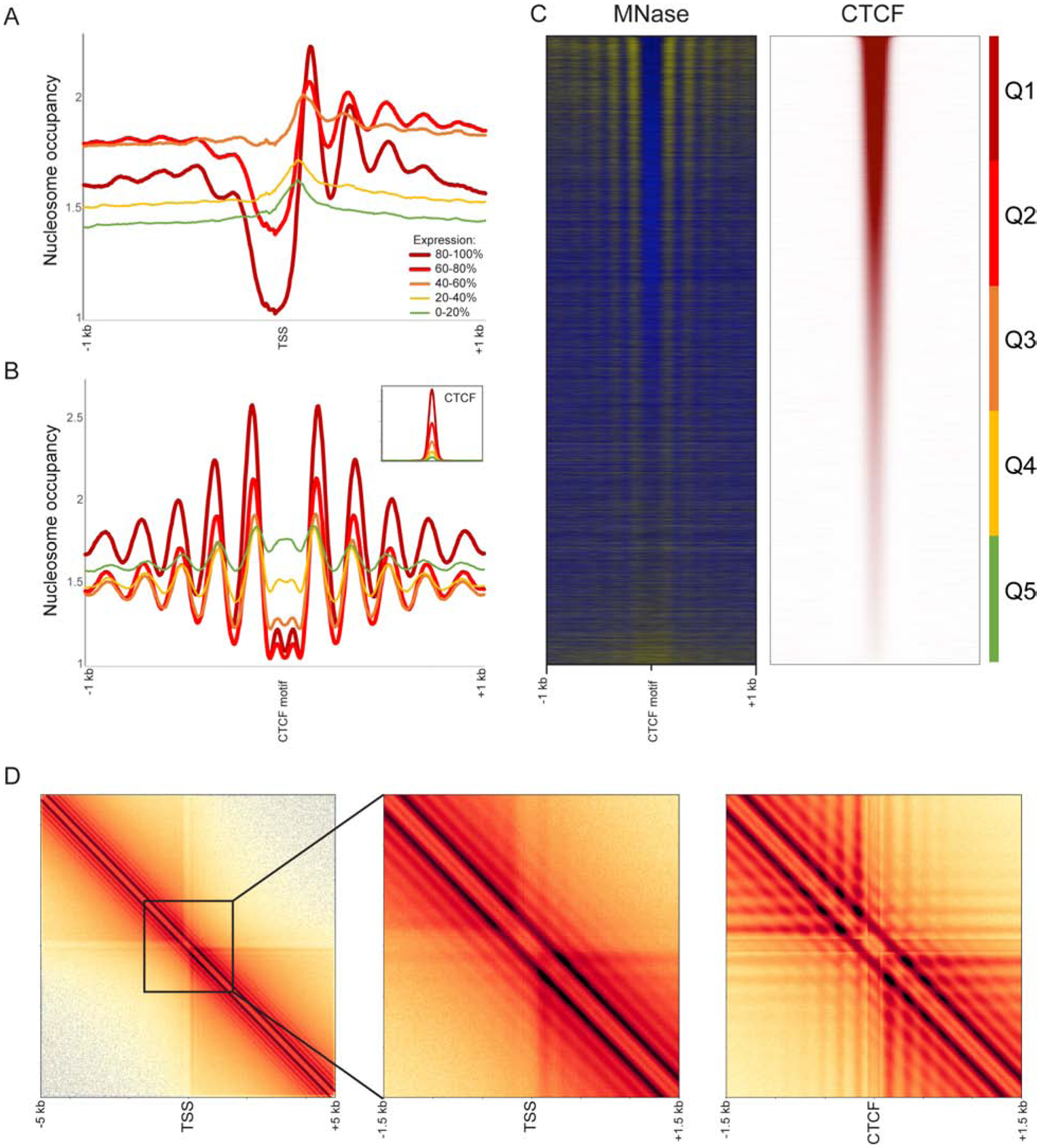
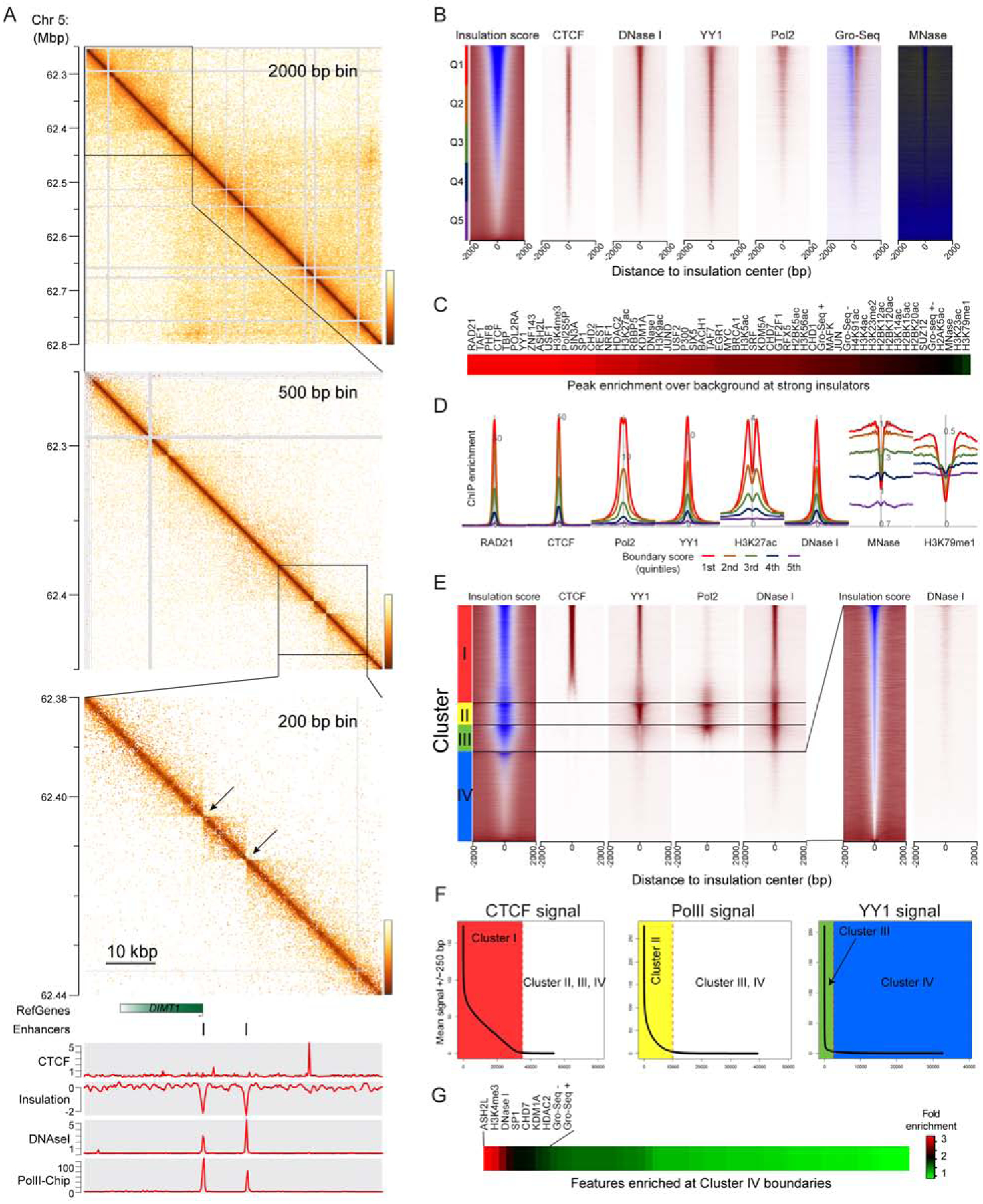
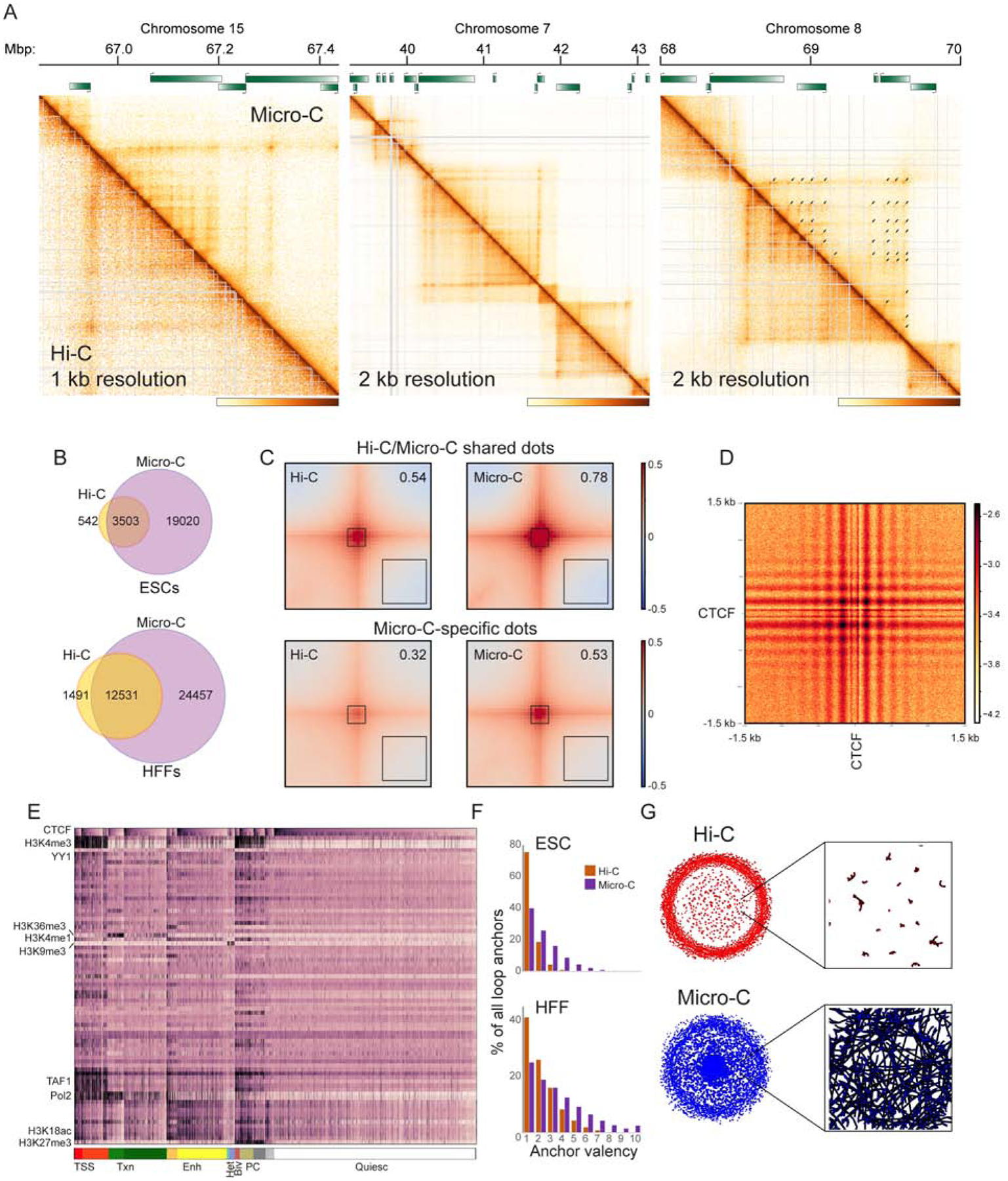
Over the past decade, 3C-related methods have provided remarkable insights into chromosome folding in vivo. To overcome the limited resolution of prior studies, we extend a recently developed Hi-C variant, Micro-C, to map chromosome architecture at nucleosome resolution in human ESCs and fibroblasts. Micro-C robustly captures known features of chromosome folding including compartment organization, topologically associating domains, and interactions between CTCF binding sites. In addition, Micro-C provides a detailed map of nucleosome positions and localizes contact domain boundaries with nucleosomal precision. Compared to Hi-C, Micro-C exhibits an order of magnitude greater dynamic range, allowing the identification of ∼20,000 additional loops in each cell type. Many newly identified peaks are localized along extrusion stripes and form transitive grids, consistent with their anchors being pause sites impeding cohesin-dependent loop extrusion. Our analyses comprise the highest-resolution maps of chromosome folding in human cells to date, providing a valuable resource for studies of chromosome organization.
Keywords: CTCF; Chromatin; Chromosomes; Loop extrusion; Micro-C.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Comment in
-
Chromosome structure at micro-scale.Nat Rev Genet. 2020 Jun;21(6):337. doi: 10.1038/s41576-020-0243-y. Nat Rev Genet. 2020. PMID: 32346116 No abstract available.
-
Of Dots and Stripes: The Morse Code of Micro-C Reveals the Ultrastructure of Transcriptional and Architectural Mammalian 3D Genome Organization.Mol Cell. 2020 May 7;78(3):376-378. doi: 10.1016/j.molcel.2020.04.021. Mol Cell. 2020. PMID: 32386540
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
