

Mylk3 null C57BL/6N mice develop cardiomyopathy, whereas Nnt null C57BL/6J mice do not
- PMID: 32213617
- PMCID: PMC7103425
- DOI: 10.26508/lsa.201900593
Mylk3 null C57BL/6N mice develop cardiomyopathy, whereas Nnt null C57BL/6J mice do not
Abstract
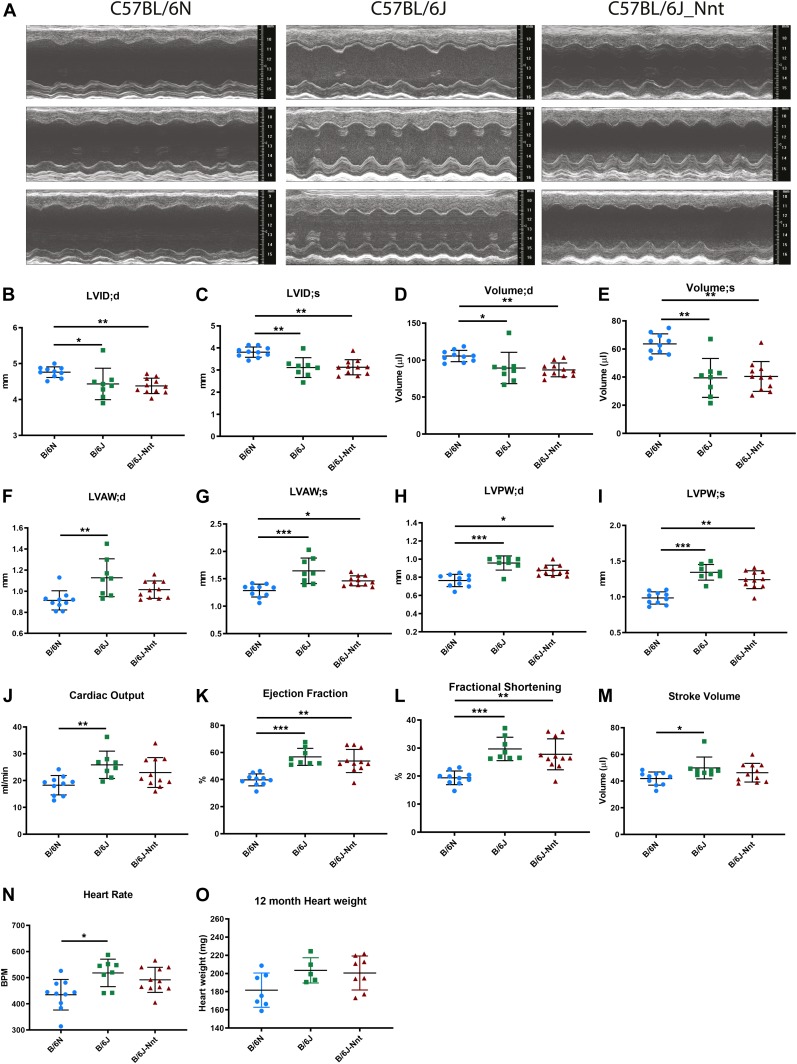
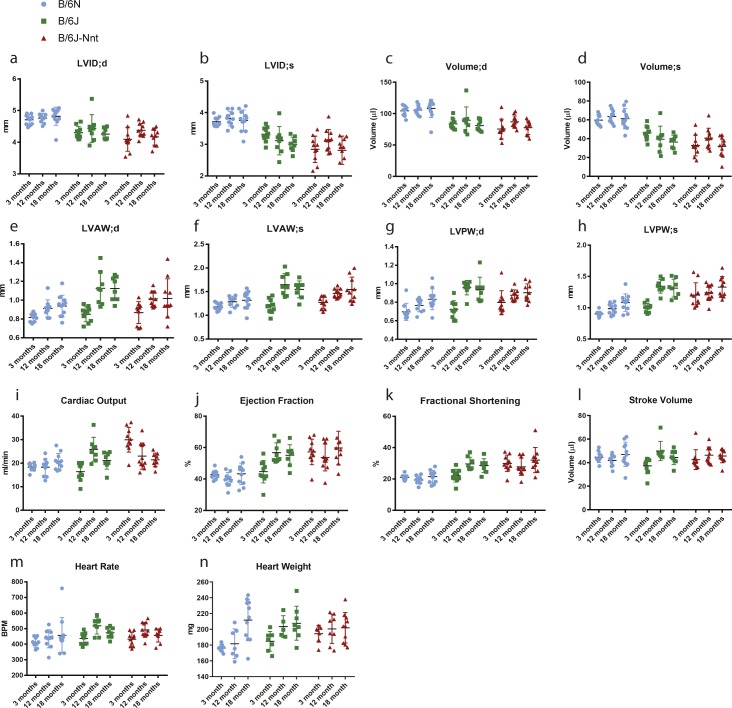
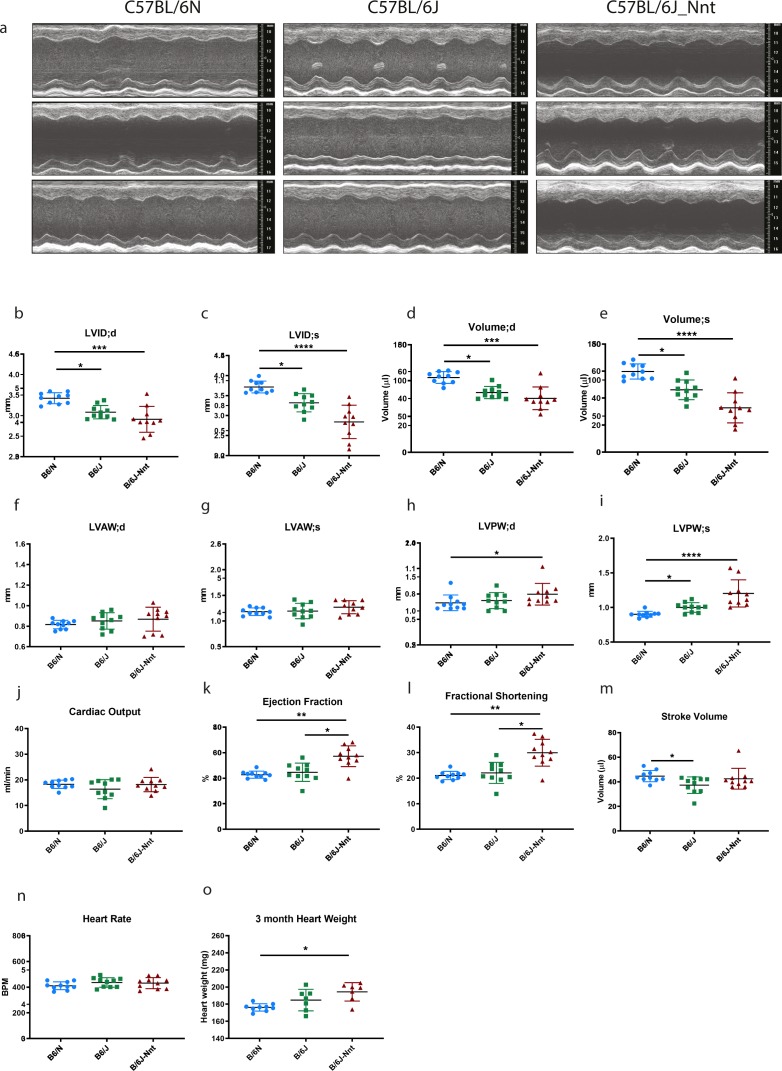
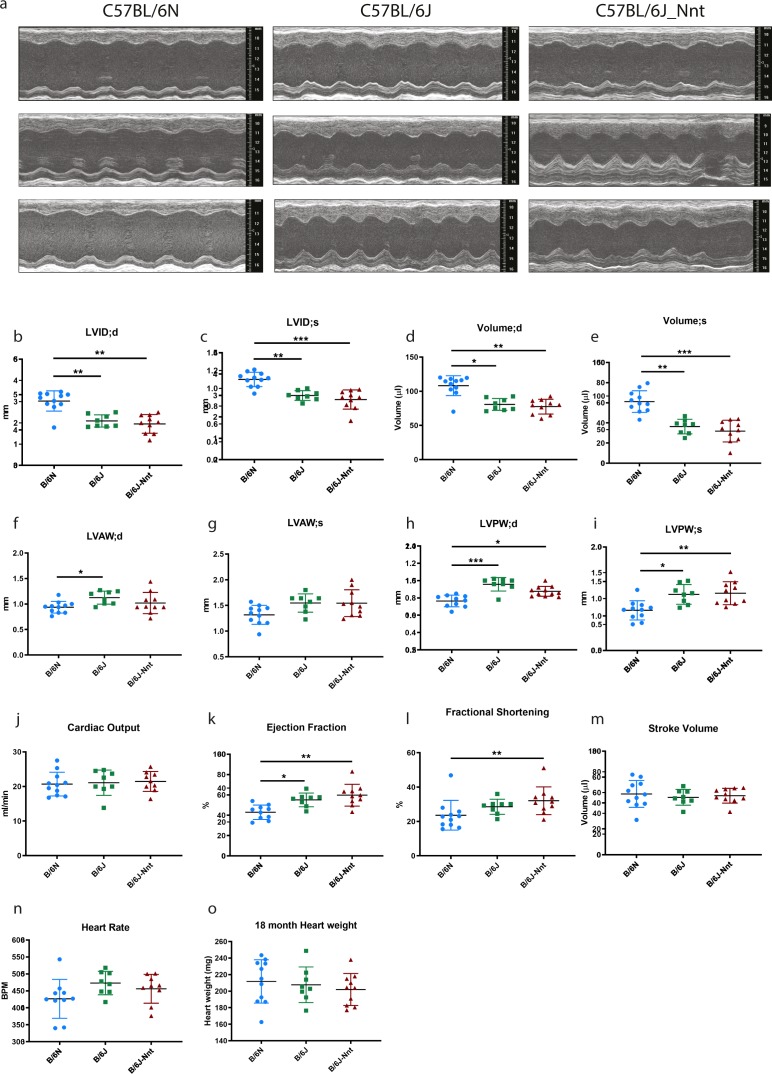
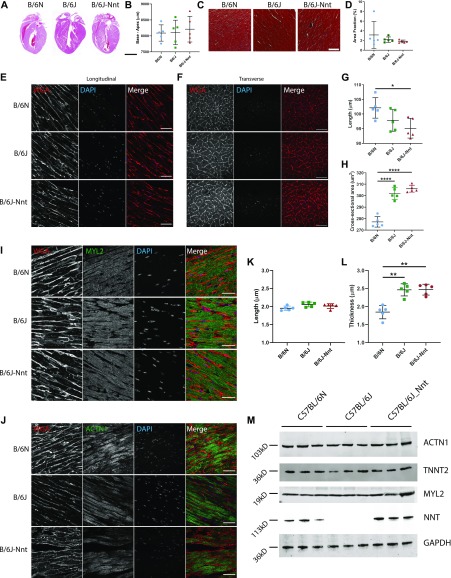
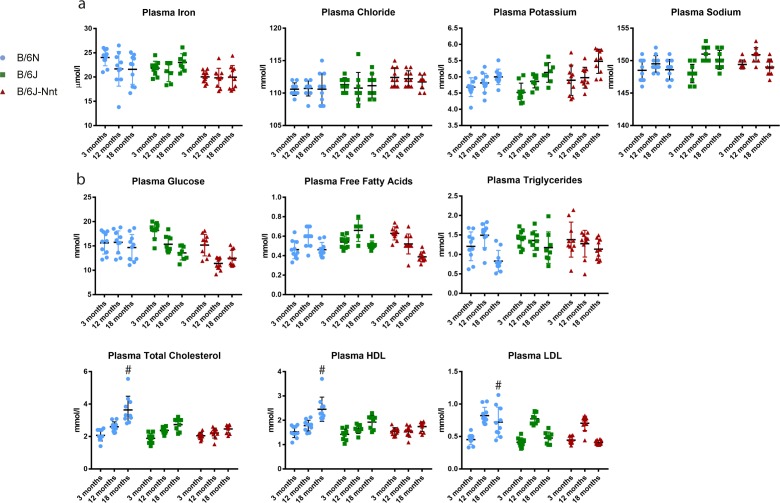
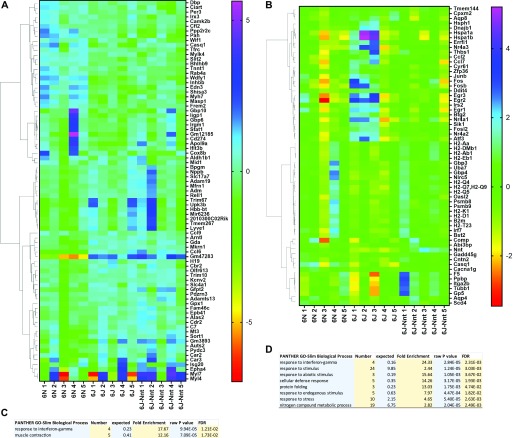
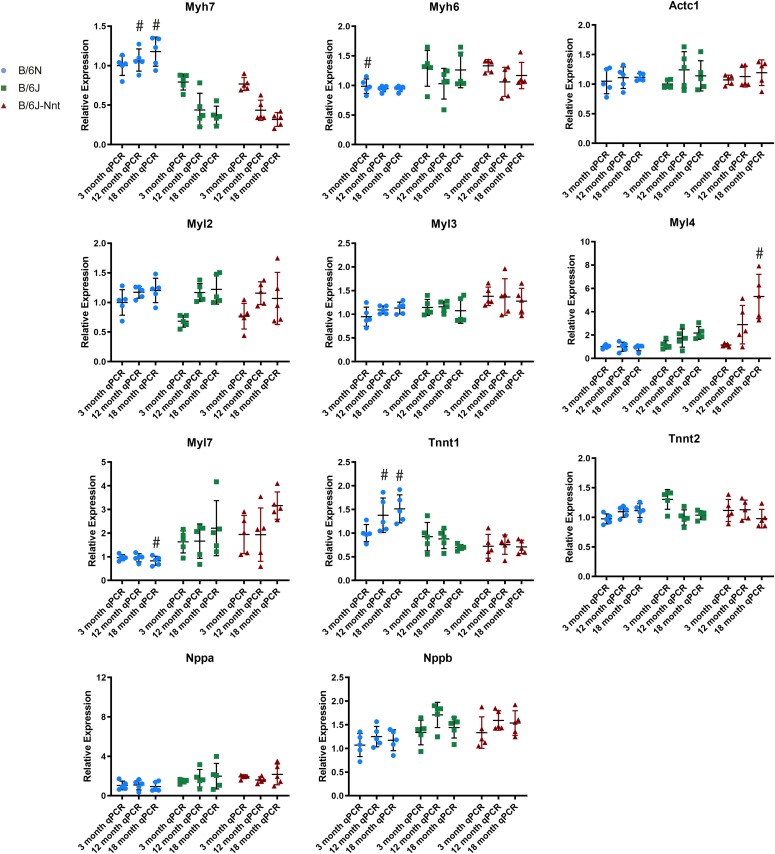
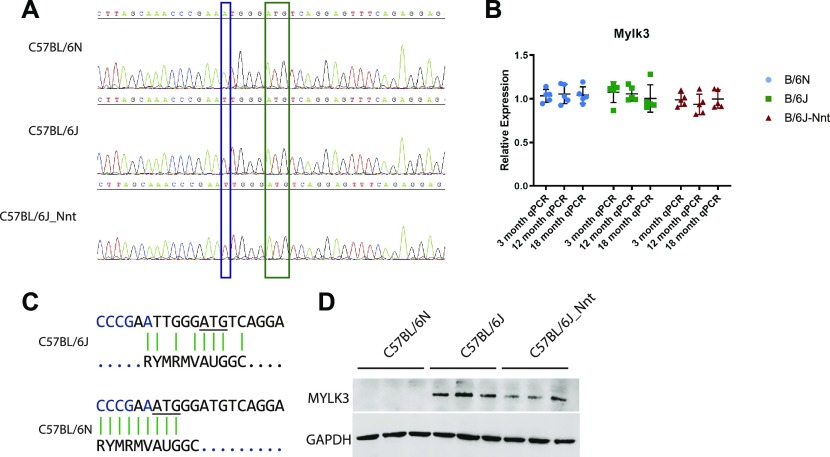
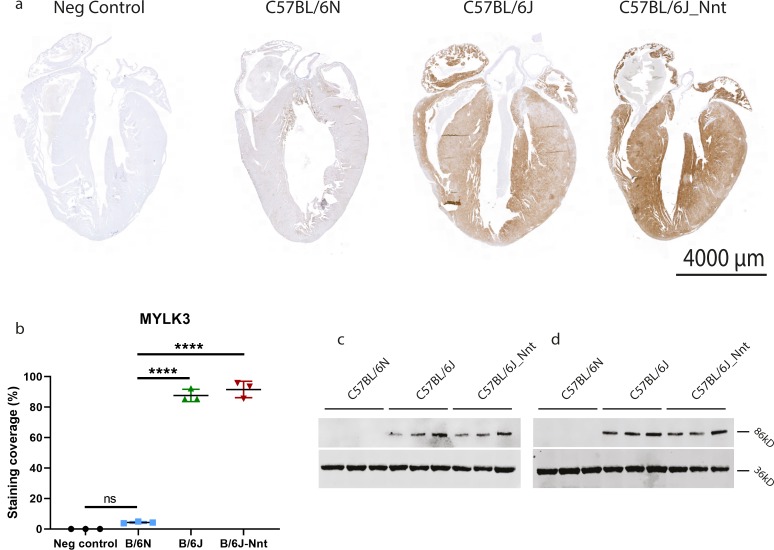
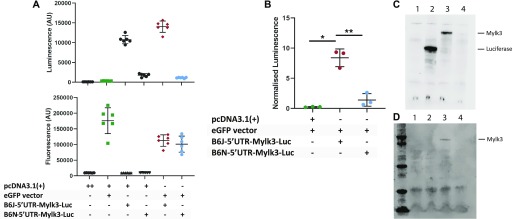
The C57BL/6J and C57BL/6N mice have well-documented phenotypic and genotypic differences, including the infamous nicotinamide nucleotide transhydrogenase (Nnt) null mutation in the C57BL/6J substrain, which has been linked to cardiovascular traits in mice and cardiomyopathy in humans. To assess whether Nnt loss alone causes a cardiovascular phenotype, we investigated the C57BL/6N, C57BL/6J mice and a C57BL/6J-BAC transgenic rescuing NNT expression, at 3, 12, and 18 mo. We identified a modest dilated cardiomyopathy in the C57BL/6N mice, absent in the two B6J substrains. Immunofluorescent staining of cardiomyocytes revealed eccentric hypertrophy in these mice, with defects in sarcomere organisation. RNAseq analysis identified differential expression of a number of cardiac remodelling genes commonly associated with cardiac disease segregating with the phenotype. Variant calling from RNAseq data identified a myosin light chain kinase 3 (Mylk3) mutation in C57BL/6N mice, which abolishes MYLK3 protein expression. These results indicate the C57BL/6J Nnt-null mice do not develop cardiomyopathy; however, we identified a null mutation in Mylk3 as a credible cause of the cardiomyopathy phenotype in the C57BL/6N.
© 2020 Williams et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases