

Identification of hub genes in hepatocellular carcinoma using integrated bioinformatic analysis
- PMID: 32213663
- PMCID: PMC7138582
- DOI: 10.18632/aging.102969
Identification of hub genes in hepatocellular carcinoma using integrated bioinformatic analysis
Abstract
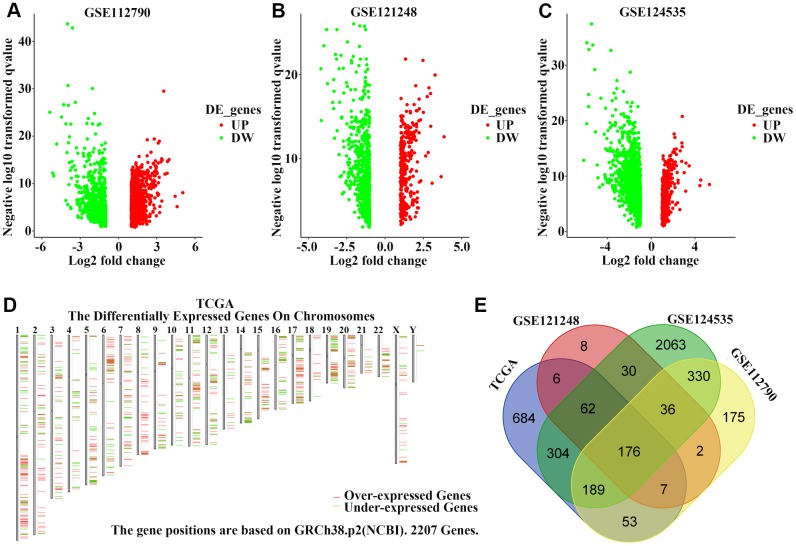
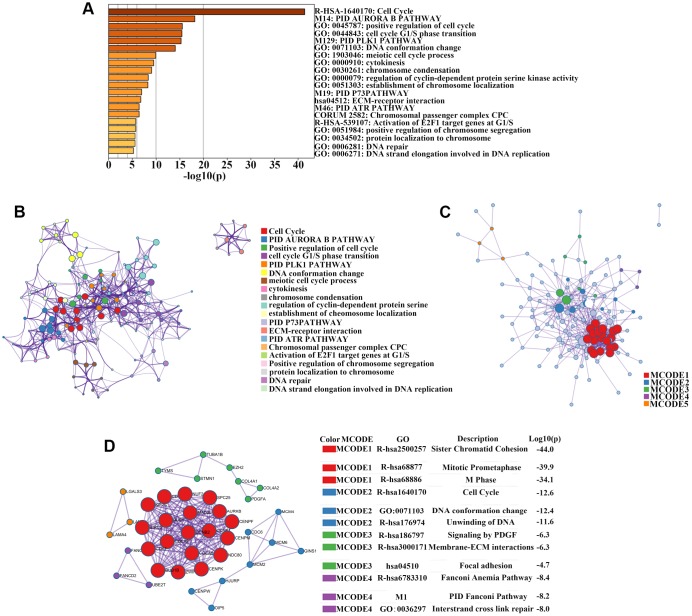
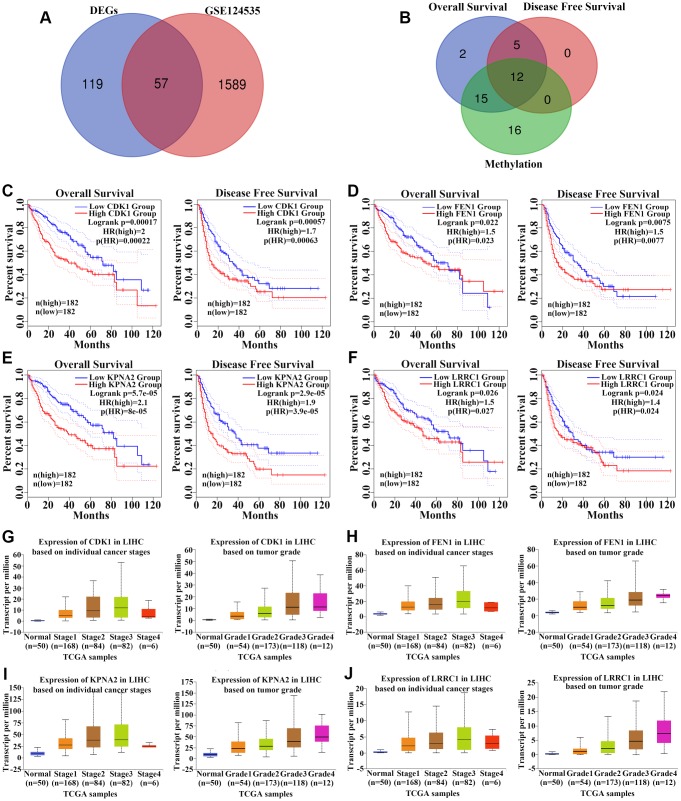
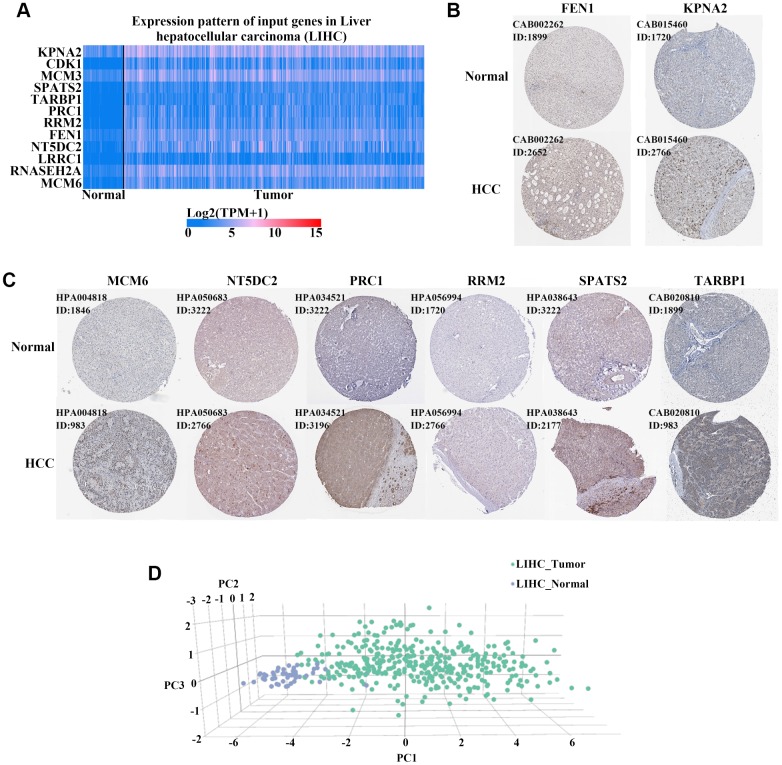
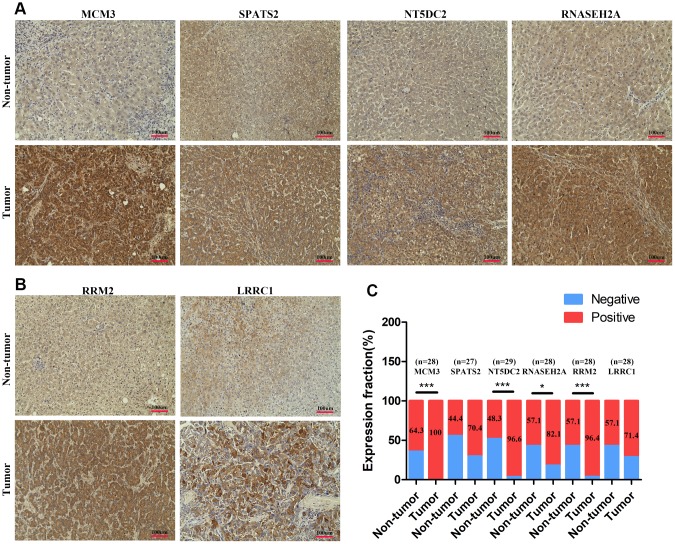
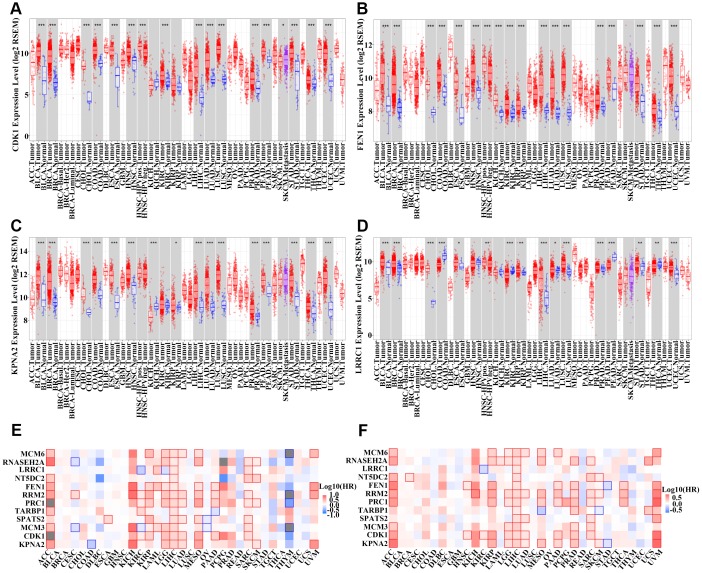
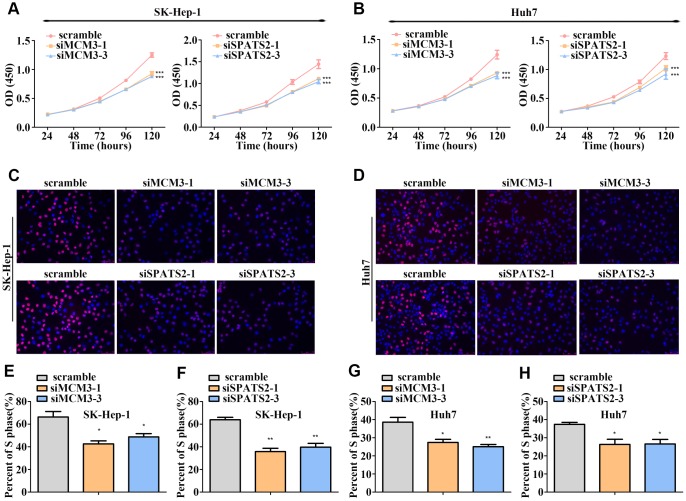
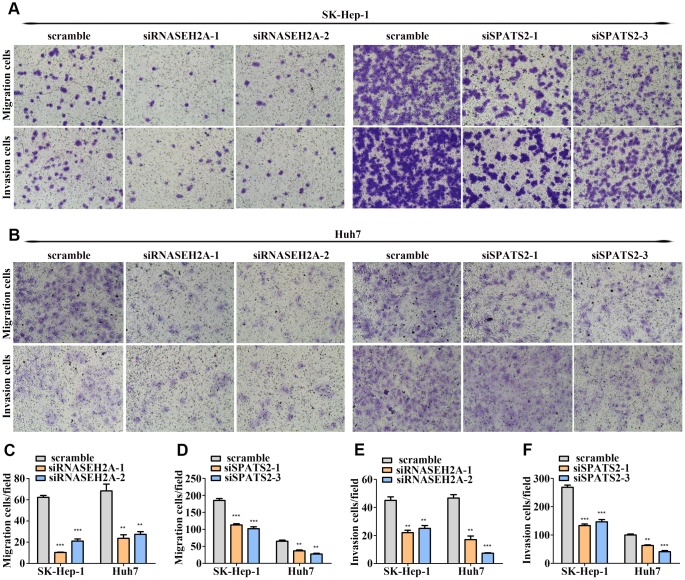
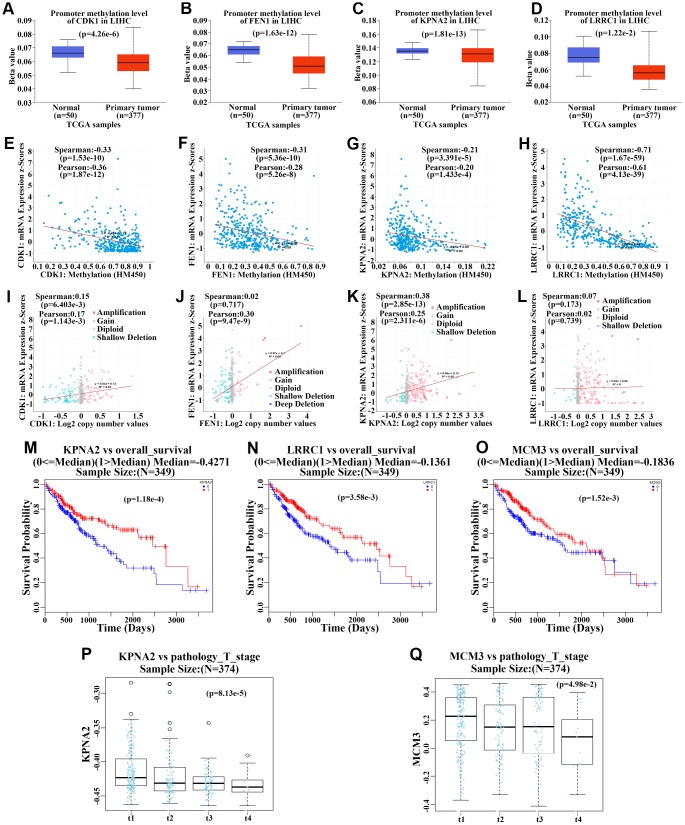
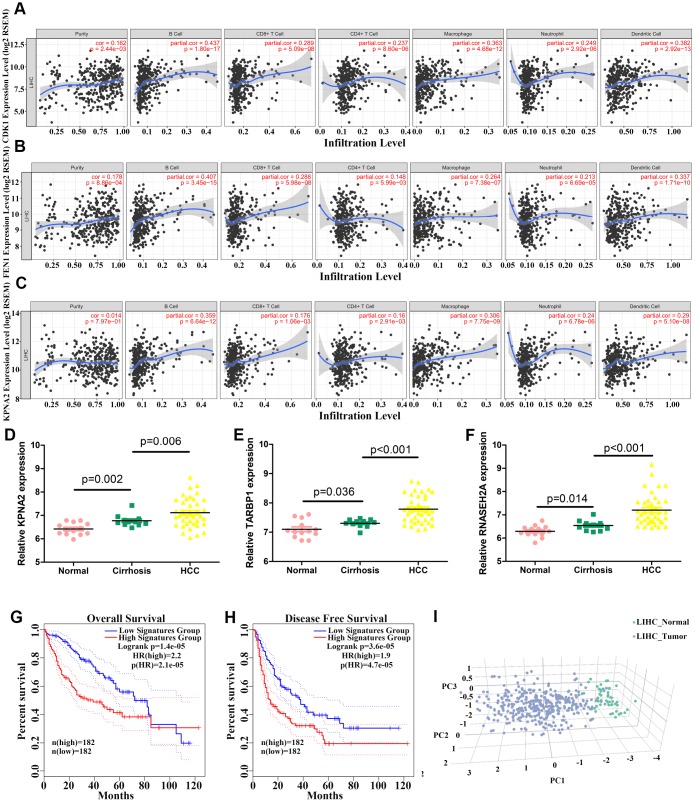
The molecular mechanisms underlying hepatocellular carcinoma (HCC) progression remain largely undefined. Here, we identified 176 commonly upregulated genes in HCC tissues based on three Gene Expression Omnibus datasets and The Cancer Genome Atlas (TCGA) cohort. We integrated survival and methylation analyses to further obtain 12 upregulated genes for validation. These genes were overexpressed in HCC tissues at the transcription and protein levels, and increased mRNA levels were related to higher tumor grades and cancer stages. The expression of all markers was negatively associated with overall and disease-free survival in HCC patients. Most of these hub genes can promote HCC proliferation and/or metastasis. These 12 hub genes were also overexpressed and had strong prognostic value in many other cancer types. Methylation and gene copy number analyses indicated that the upregulation of these hub genes was probably due to hypomethylation or increased gene copy numbers. Further, the methylation levels of three genes, KPNA2, MCM3, and LRRC1, were associated with HCC clinical features. Moreover, the levels of most hub genes were related to immune cell infiltration in HCC microenvironments. Finally, we identified three upregulated genes (KPNA2, TARBP1, and RNASEH2A) that could comprehensively and accurately provide diagnostic and prognostic value for HCC patients.
Keywords: bioinformatic analysis; diagnostic value; hepatocellular carcinoma; hub genes; prognostic value.
Conflict of interest statement
Figures
References
-
- Ni M, Liu X, Wu J, Zhang D, Tian J, Wang T, Liu S, Meng Z, Wang K, Duan X, Zhou W, Zhang X. Identification of candidate biomarkers correlated with the pathogenesis and prognosis of non-small cell lung cancer via integrated bioinformatics analysis. Front Genet. 2018; 9:469. 10.3389/fgene.2018.00469 - DOI - PMC - PubMed
-
- Hou Q, Bing ZT, Hu C, Li MY, Yang KH, Mo Z, Xie XW, Liao JL, Lu Y, Horie S, Lou MW. RankProd combined with genetic algorithm optimized artificial neural network establishes a diagnostic and prognostic prediction model that revealed C1QTNF3 as a biomarker for prostate cancer. EBioMedicine. 2018; 32:234–44. 10.1016/j.ebiom.2018.05.010 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
