

Viruses with different genome types adopt a similar strategy to pack nucleic acids based on positively charged protein domains
- PMID: 32214181
- PMCID: PMC7096446
- DOI: 10.1038/s41598-020-62328-w
Viruses with different genome types adopt a similar strategy to pack nucleic acids based on positively charged protein domains
Abstract
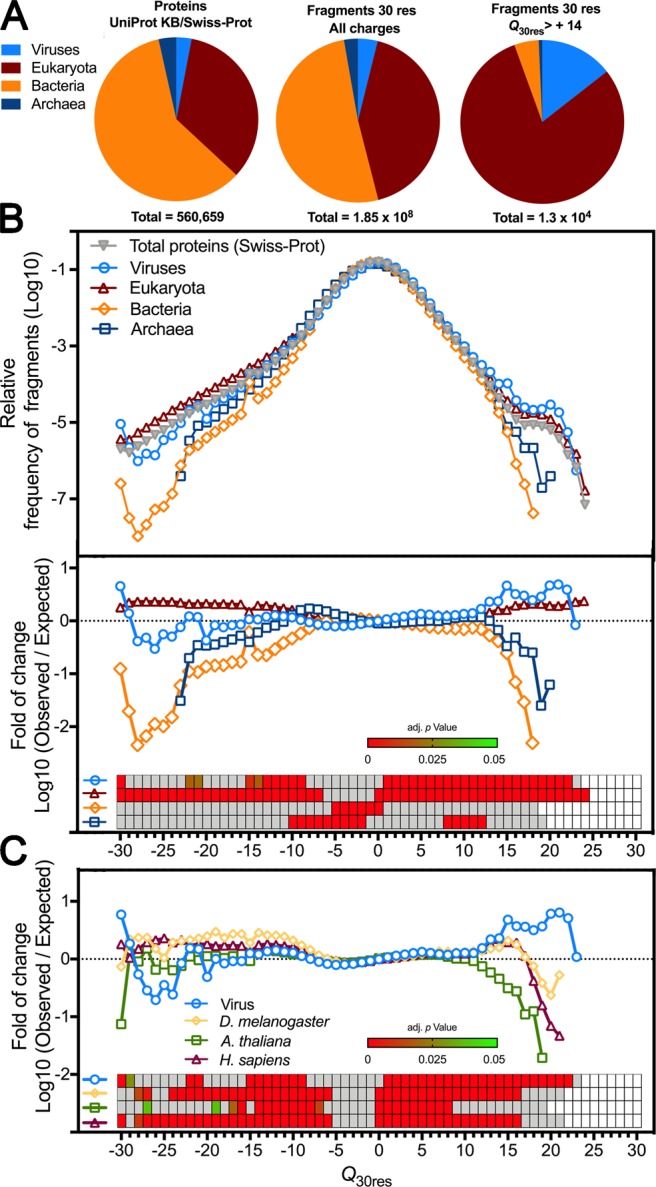
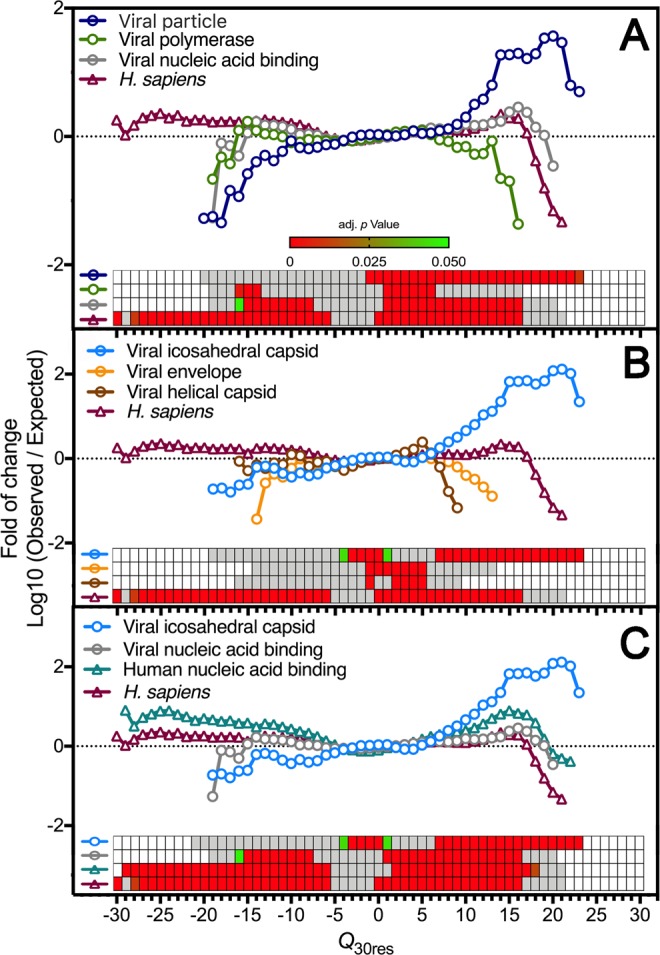
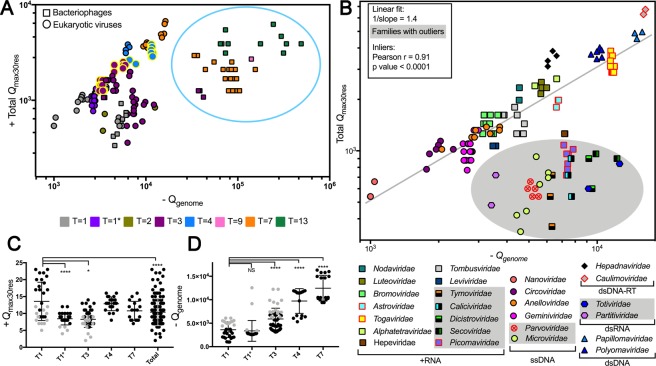
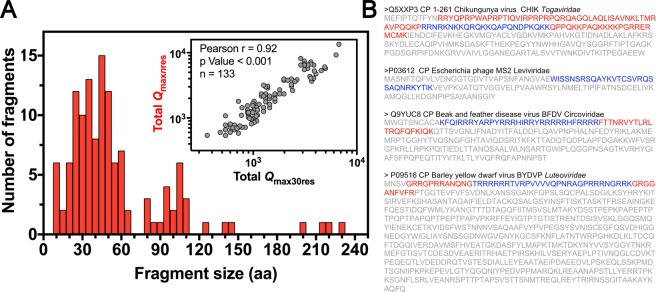
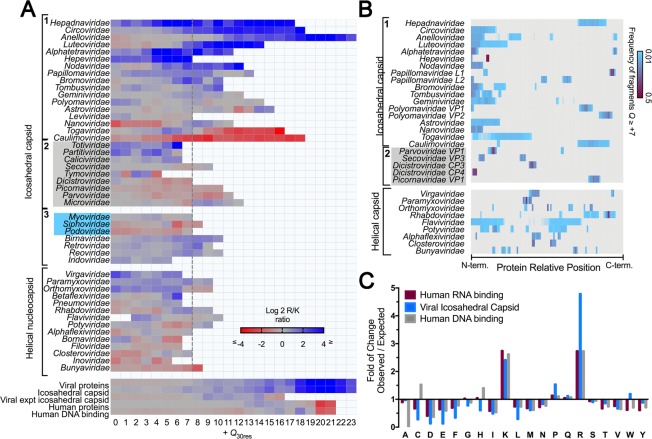
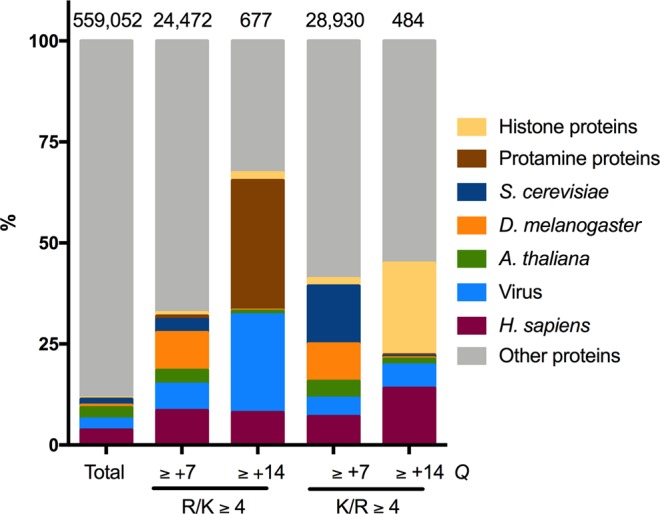
Capsid proteins often present a positively charged arginine-rich sequence at their terminal regions, which has a fundamental role in genome packaging and particle stability for some icosahedral viruses. These sequences show little to no conservation and are structurally dynamic such that they cannot be easily detected by common sequence or structure comparisons. As a result, the occurrence and distribution of positively charged domains across the viral universe are unknown. Based on the net charge calculation of discrete protein segments, we identified proteins containing amino acid stretches with a notably high net charge (Q > + 17), which are enriched in icosahedral viruses with a distinctive bias towards arginine over lysine. We used viral particle structural data to calculate the total electrostatic charge derived from the most positively charged protein segment of capsid proteins and correlated these values with genome charges arising from the phosphates of each nucleotide. We obtained a positive correlation (r = 0.91, p-value <0001) for a group of 17 viral families, corresponding to 40% of all families with icosahedral structures described to date. These data indicated that unrelated viruses with diverse genome types adopt a common underlying mechanism for capsid assembly based on R-arms.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
