

Microbiome analyses of blood and tissues suggest cancer diagnostic approach
- PMID: 32214244
- PMCID: PMC7500457
- DOI: 10.1038/s41586-020-2095-1
Microbiome analyses of blood and tissues suggest cancer diagnostic approach
Retraction in
-
Retraction Note: Microbiome analyses of blood and tissues suggest cancer diagnostic approach.Nature. 2024 Jul;631(8021):694. doi: 10.1038/s41586-024-07656-x. Nature. 2024. PMID: 38926587 No abstract available.
Abstract
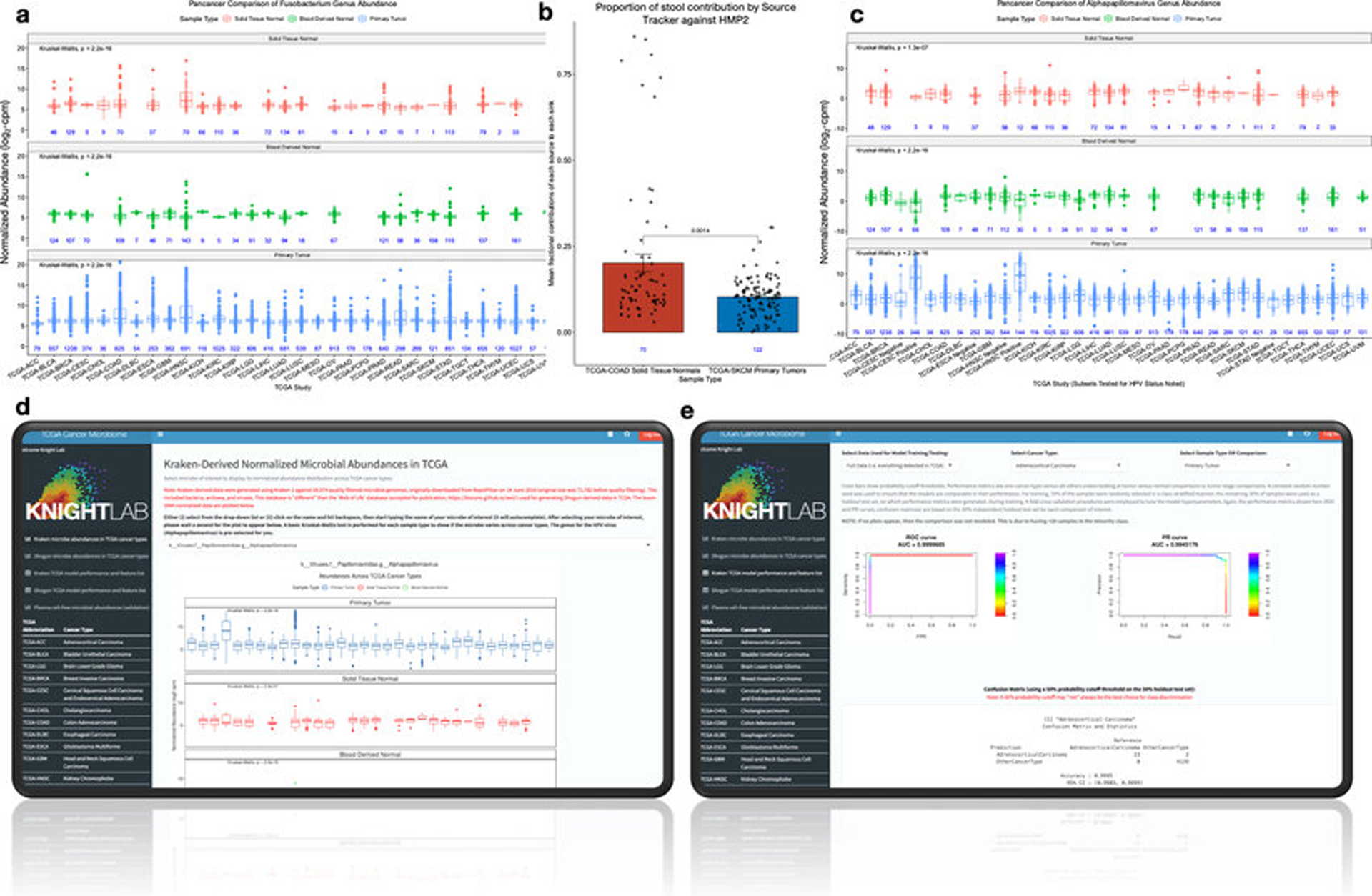
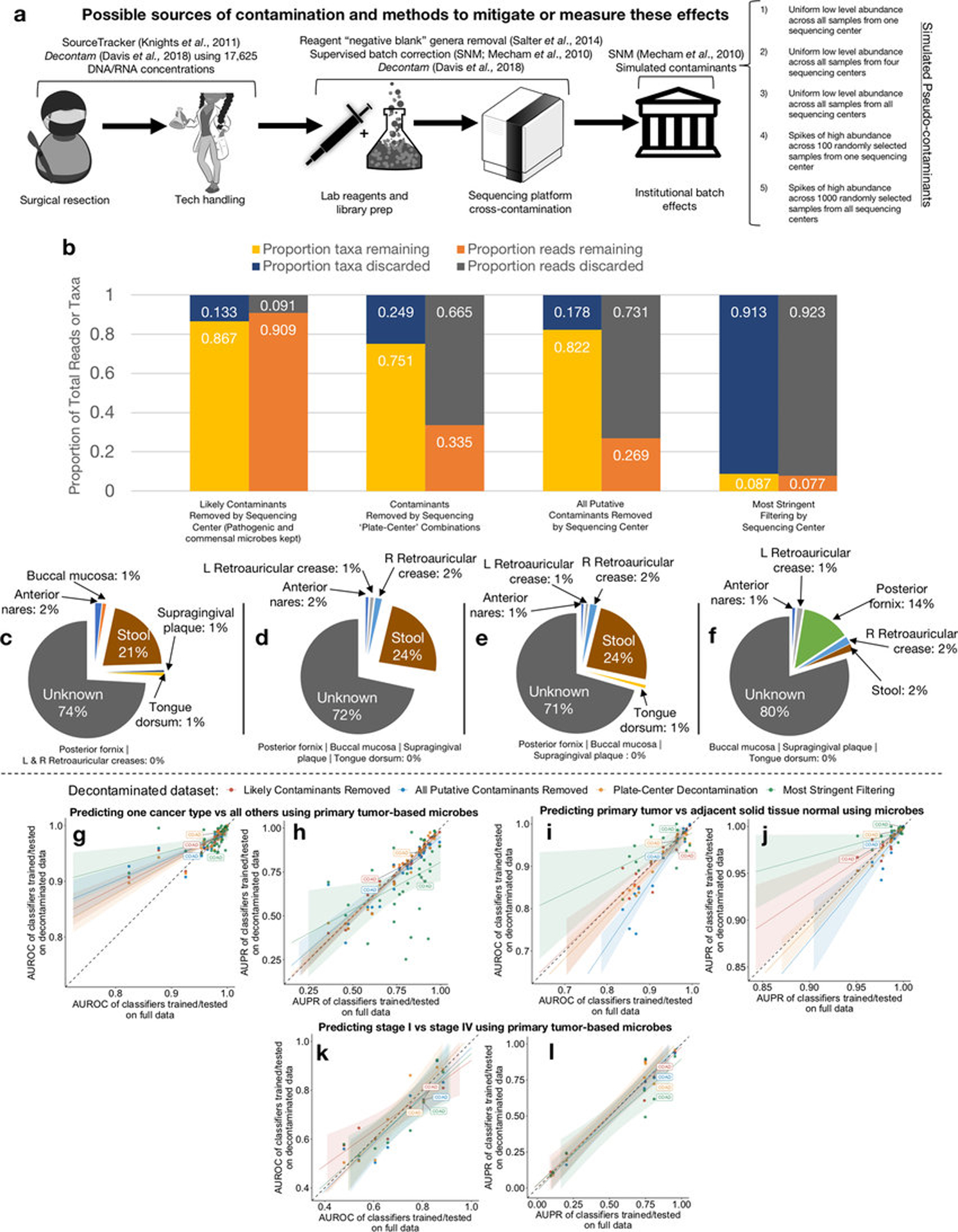
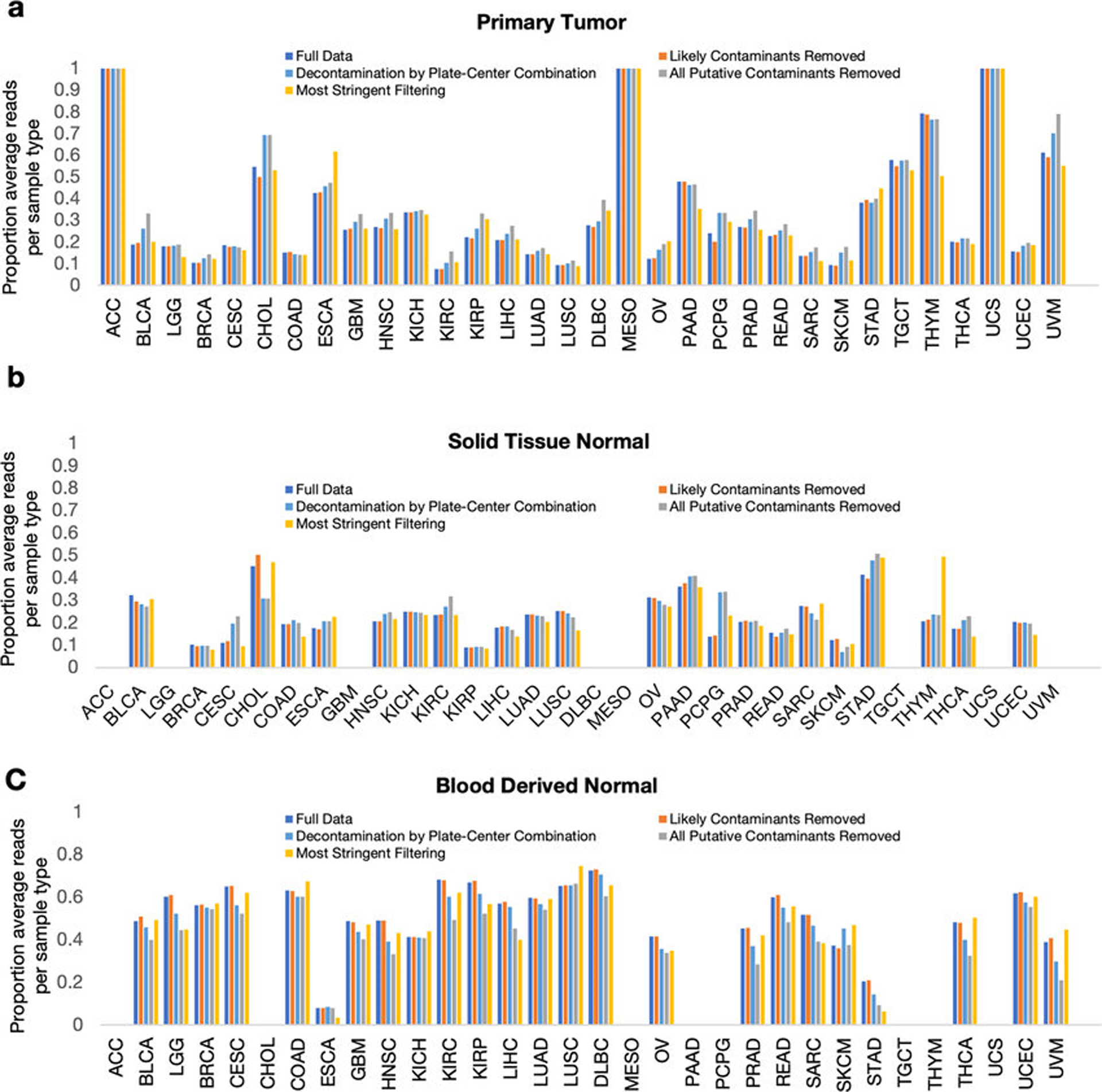
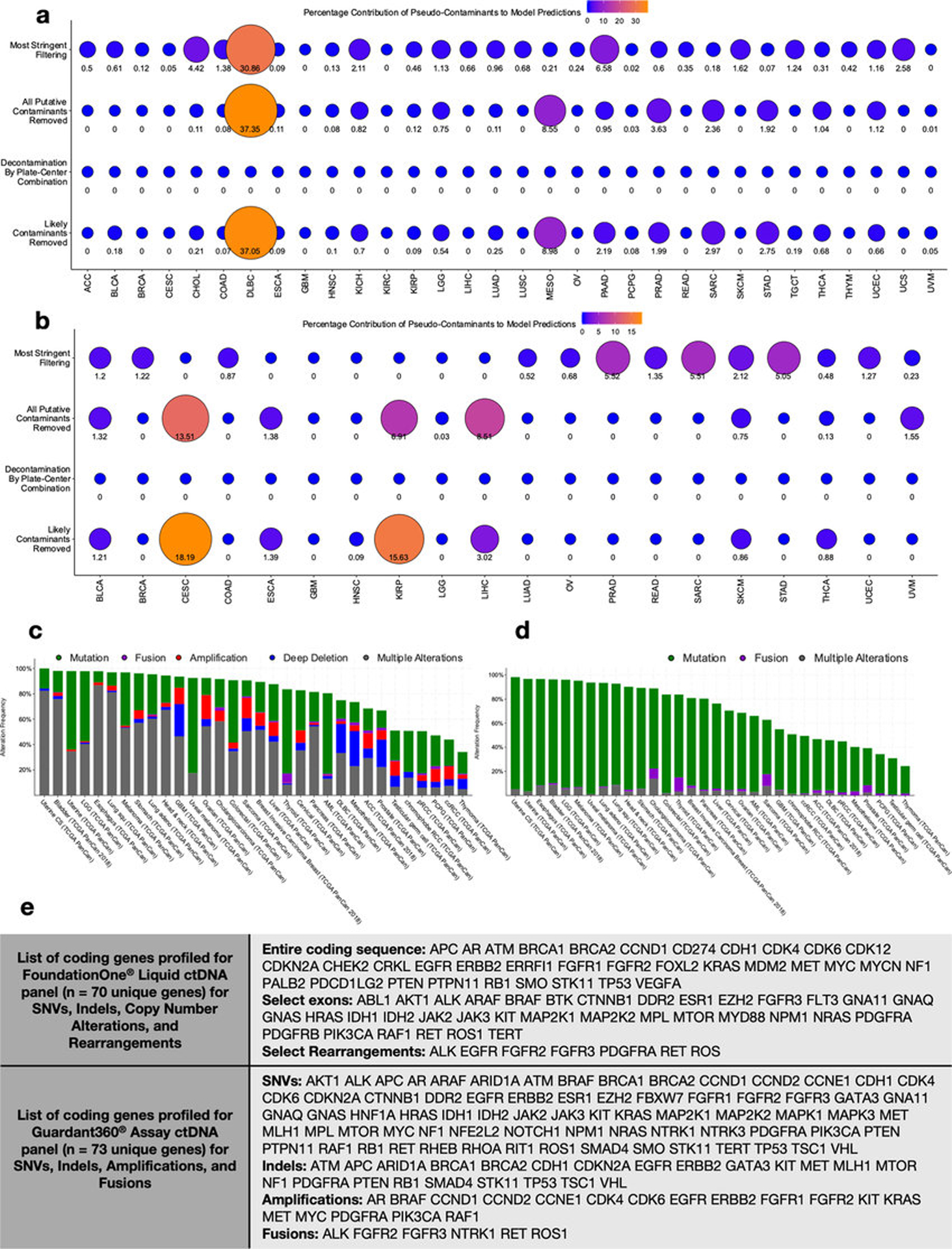
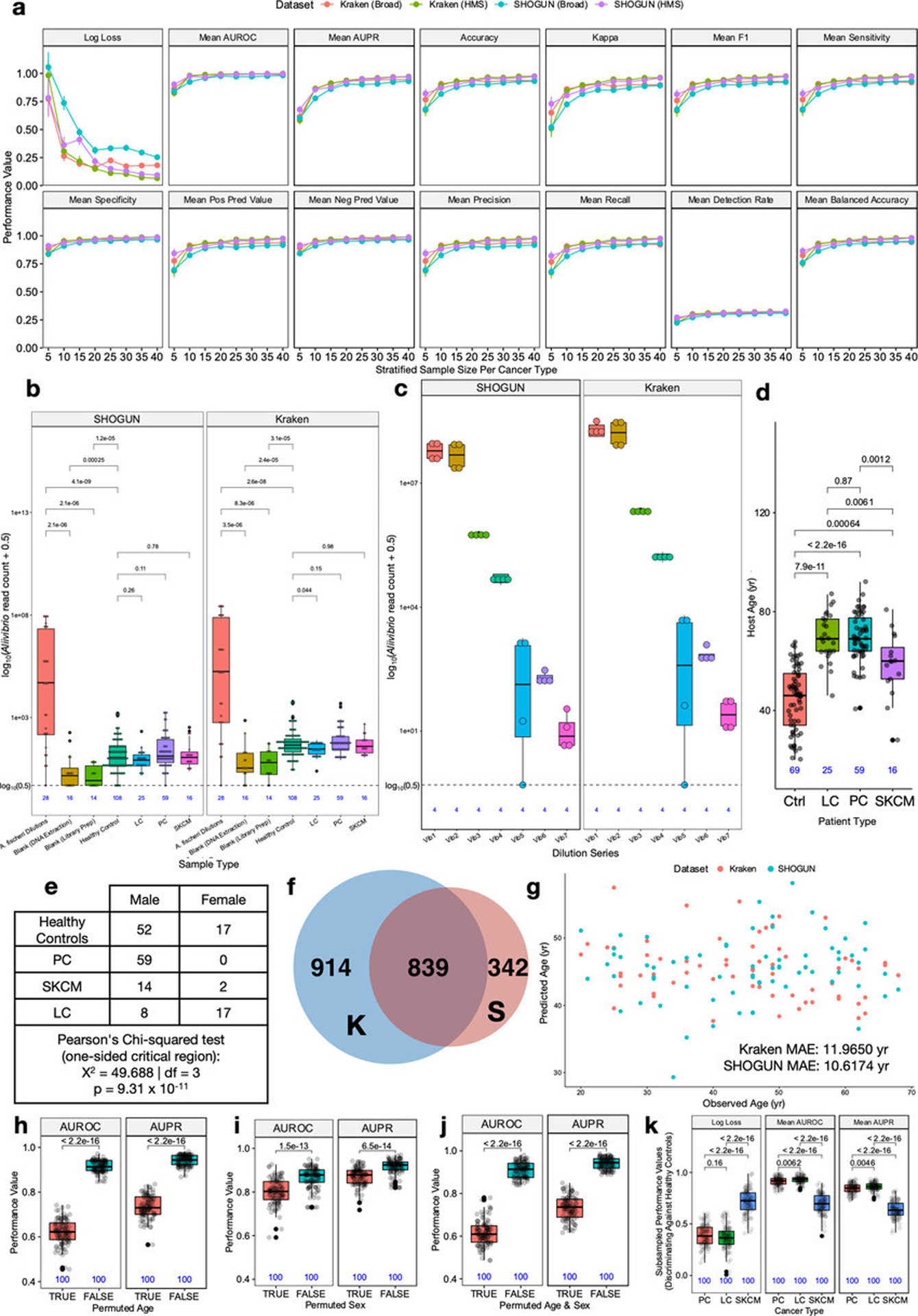
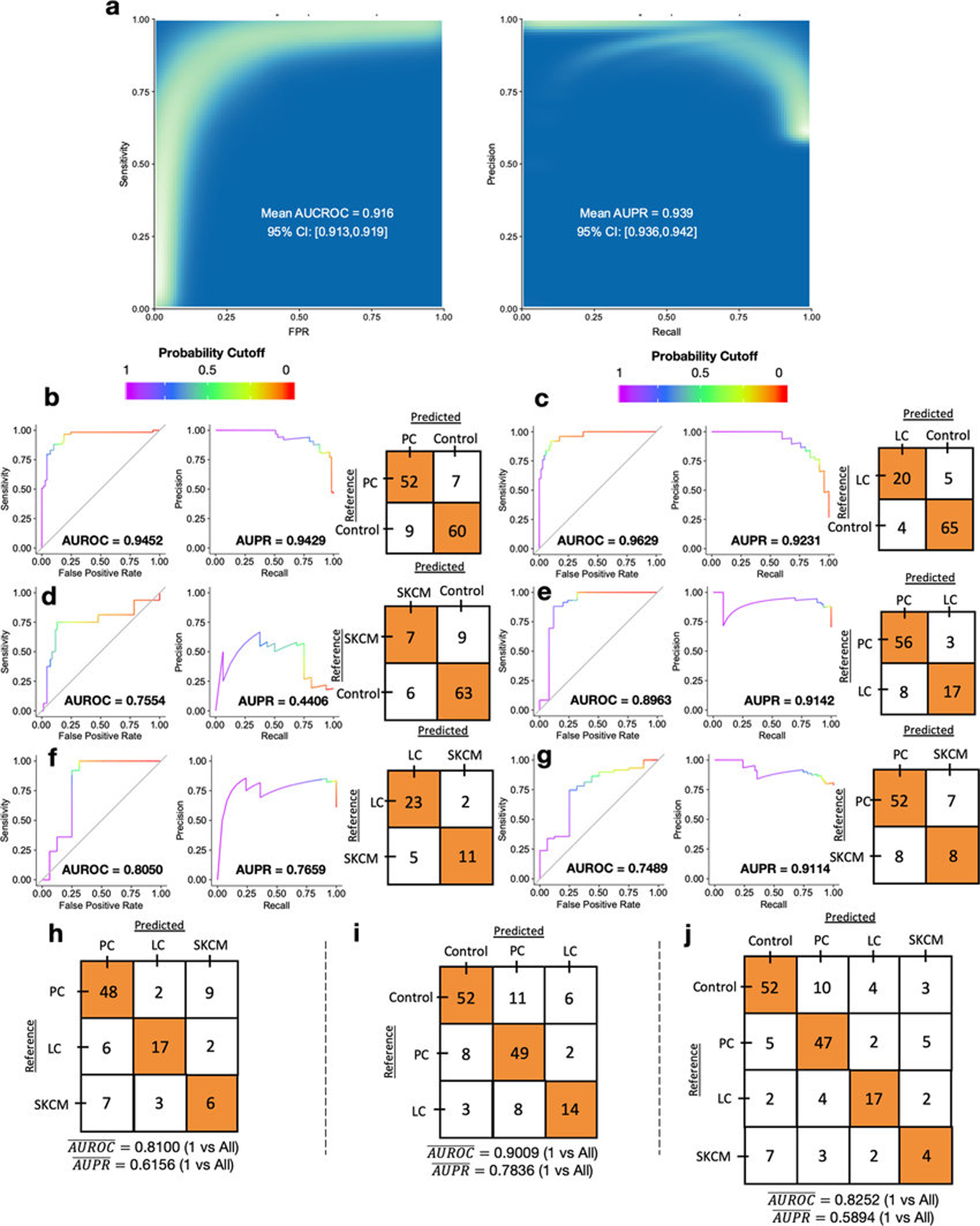
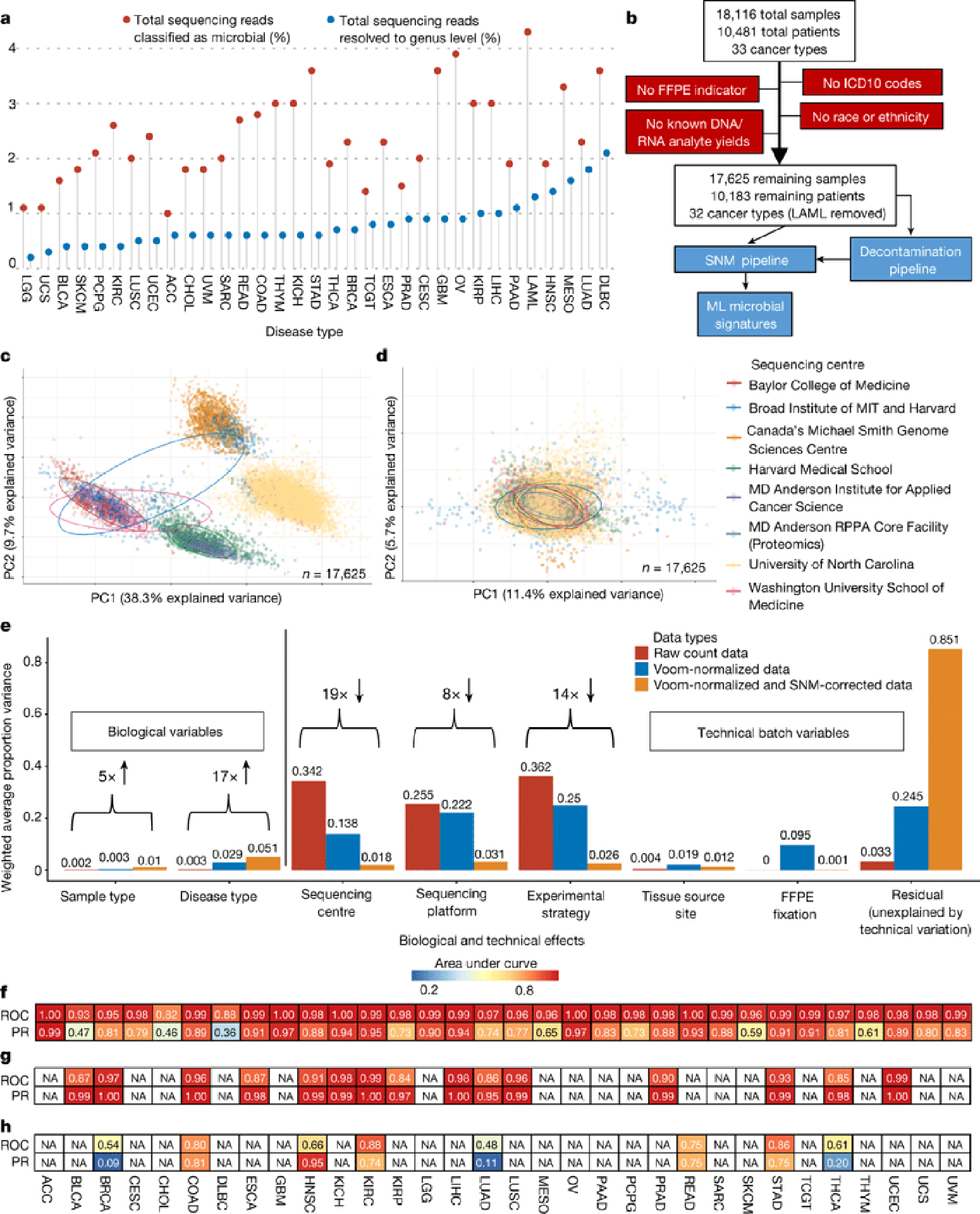
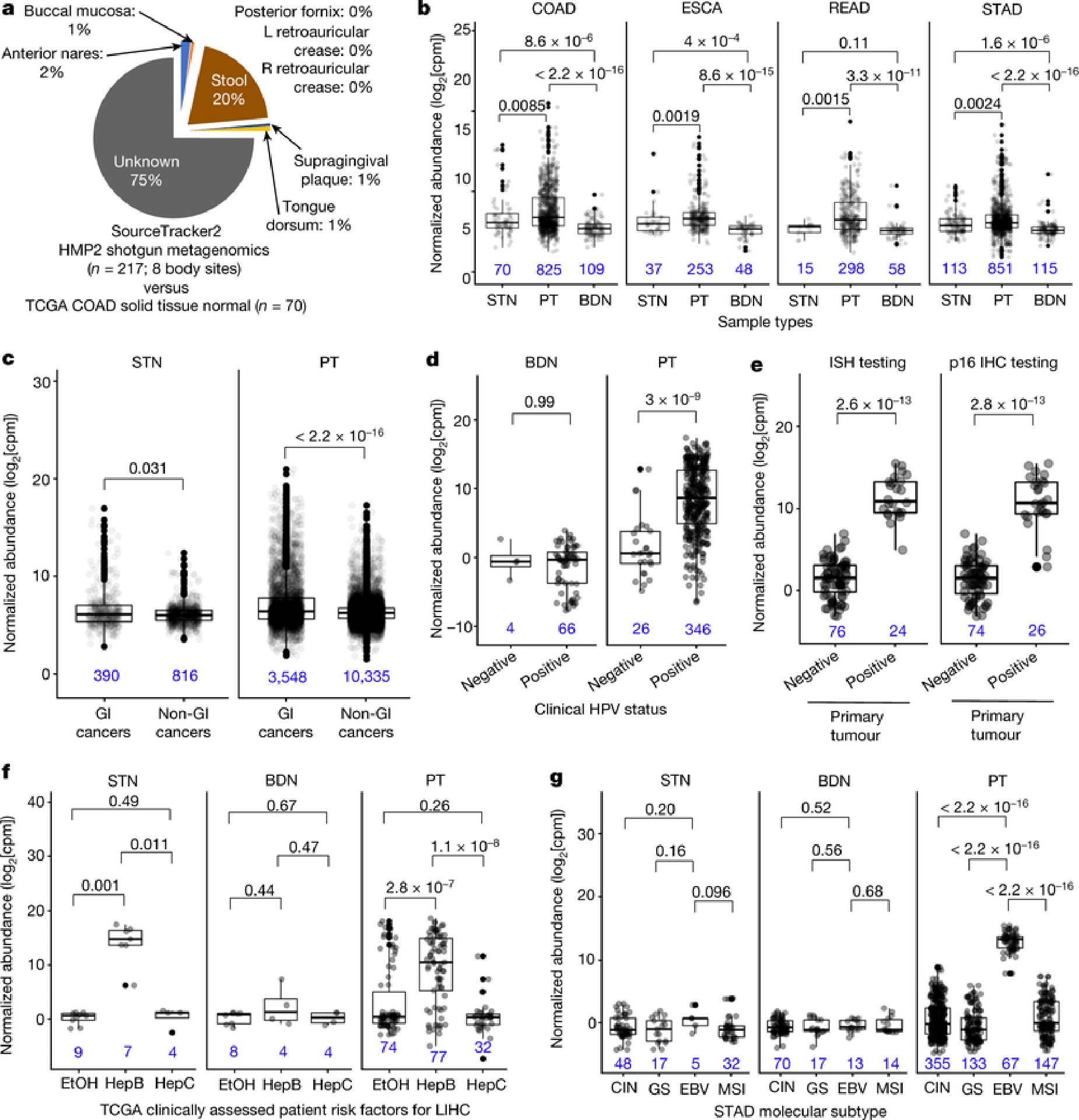
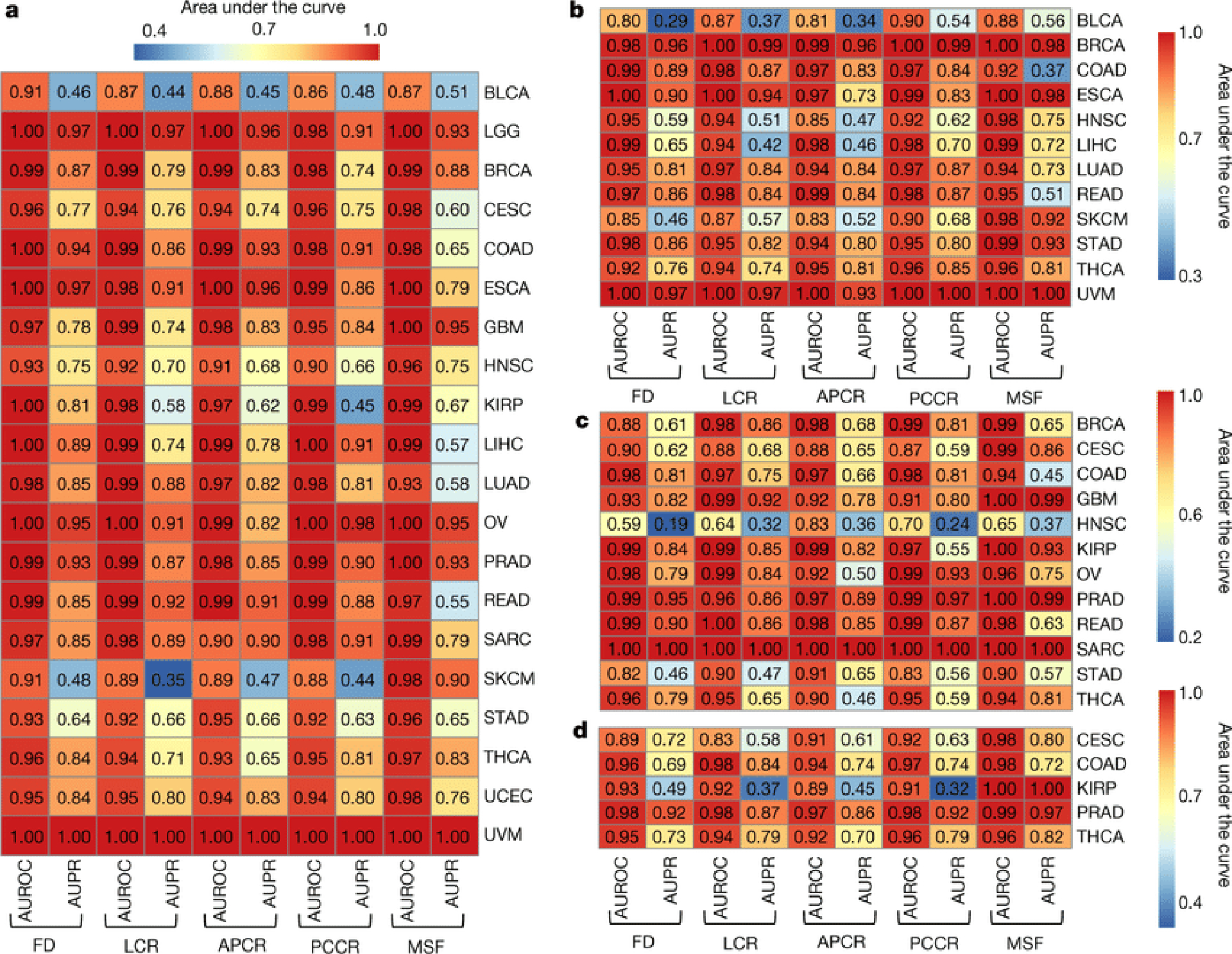
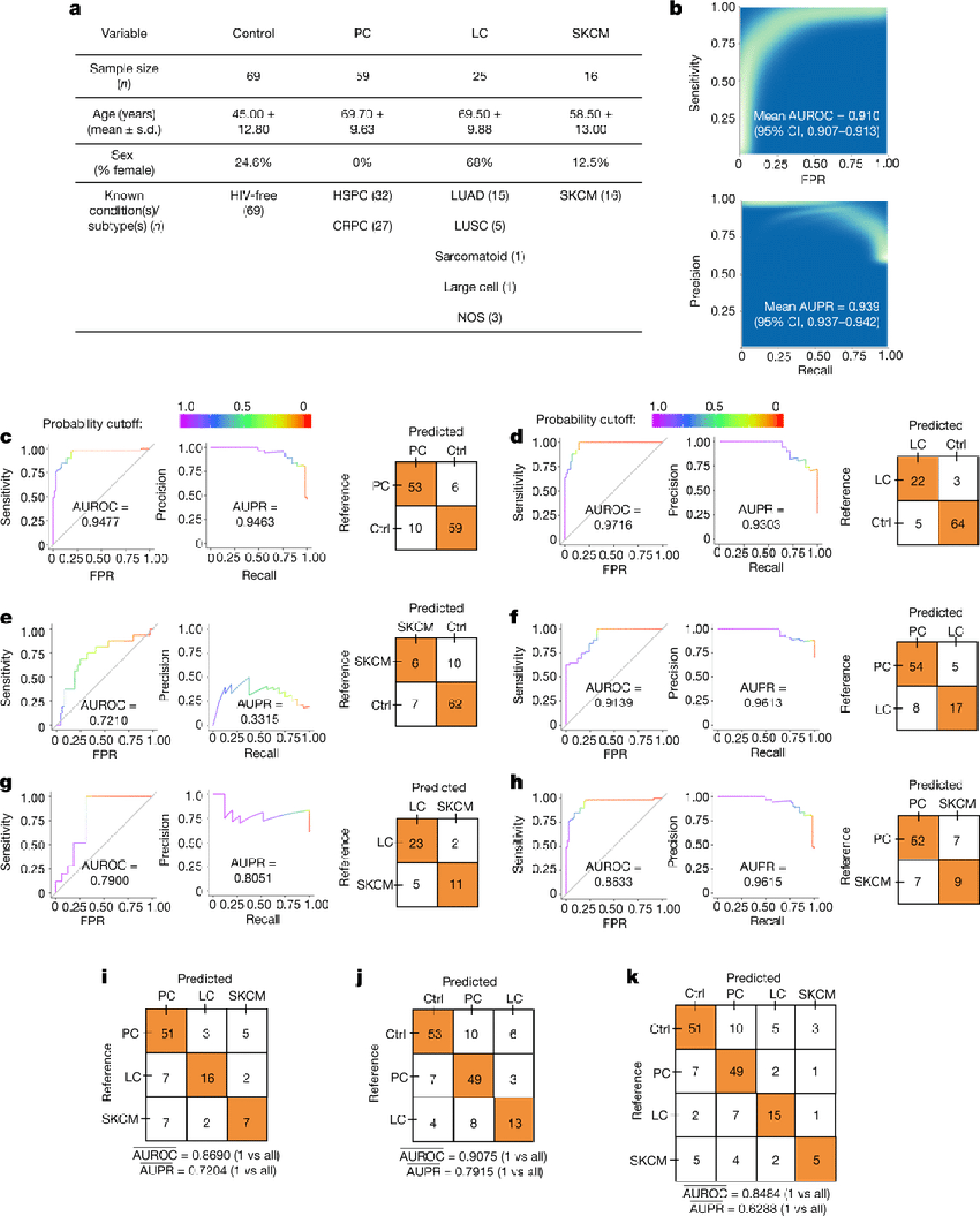
Systematic characterization of the cancer microbiome provides the opportunity to develop techniques that exploit non-human, microorganism-derived molecules in the diagnosis of a major human disease. Following recent demonstrations that some types of cancer show substantial microbial contributions1-10, we re-examined whole-genome and whole-transcriptome sequencing studies in The Cancer Genome Atlas11 (TCGA) of 33 types of cancer from treatment-naive patients (a total of 18,116 samples) for microbial reads, and found unique microbial signatures in tissue and blood within and between most major types of cancer. These TCGA blood signatures remained predictive when applied to patients with stage Ia-IIc cancer and cancers lacking any genomic alterations currently measured on two commercial-grade cell-free tumour DNA platforms, despite the use of very stringent decontamination analyses that discarded up to 92.3% of total sequence data. In addition, we could discriminate among samples from healthy, cancer-free individuals (n = 69) and those from patients with multiple types of cancer (prostate, lung, and melanoma; 100 samples in total) solely using plasma-derived, cell-free microbial nucleic acids. This potential microbiome-based oncology diagnostic tool warrants further exploration.
Conflict of interest statement
Competing interests
Clarity Genomics, the employer of E.K., did not provide funding for this study. Both G.D.P. and R.K. have jointly filed U.S. Provisional Patent Application Serial No. 62/754,696 and International Application No. PCT/US19/59647 on the basis of this work. G.D.P., R.K., and S.M.M. have started a company to commercialize the intellectual property. R.K. is a member of the SAB for GenCirq, Inc., holds an equity interest in GenCirq, and can receive reimbursements for expenses up to $5,000/yr. R.K., A.D.S., and S.M.M. are directors at the Center for Microbiome Innovation at UC San Diego, which receives industry research funding for various microbiome initiatives, but no industry funding was provided for this cancer microbiome project.
Figures
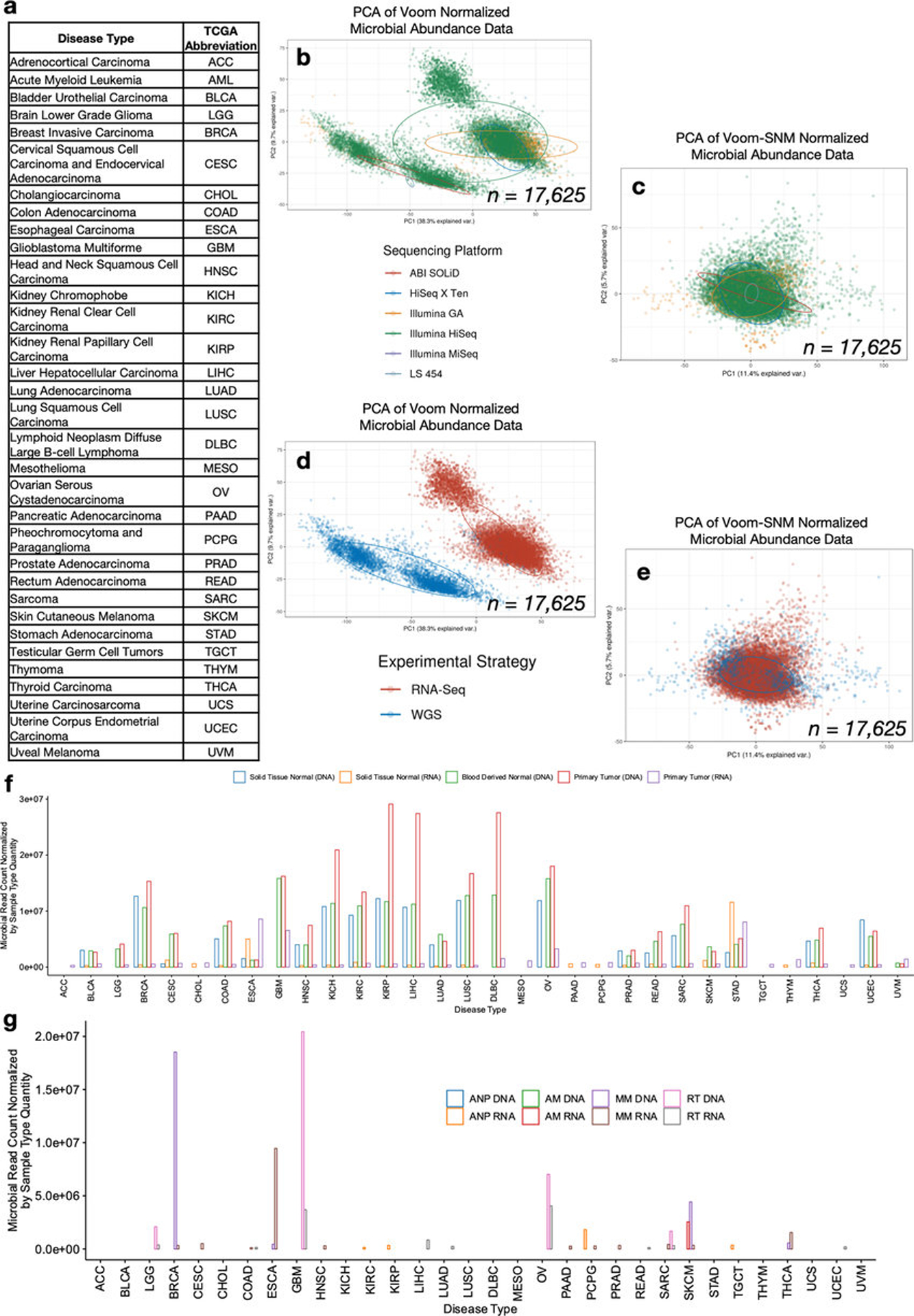
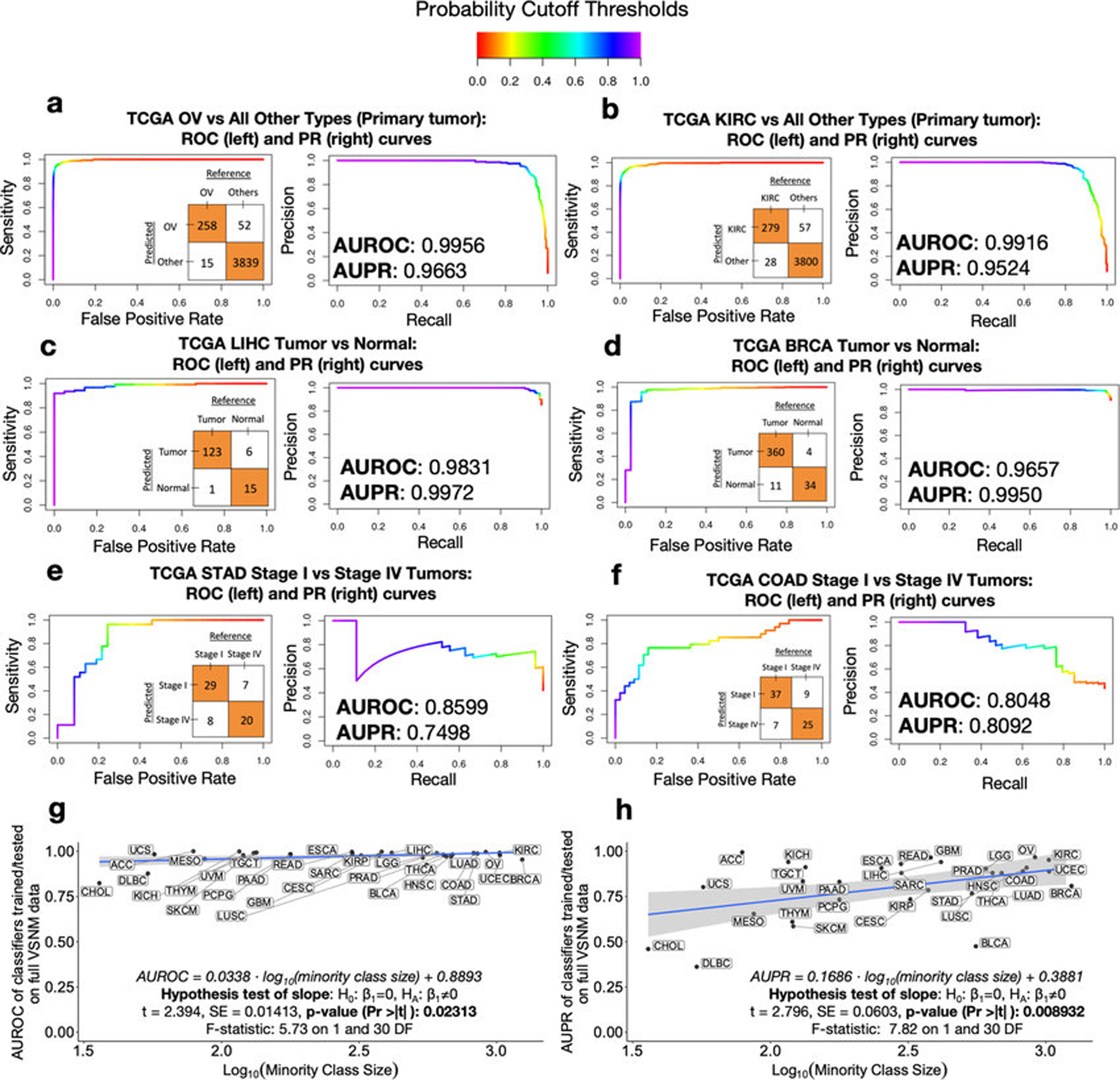
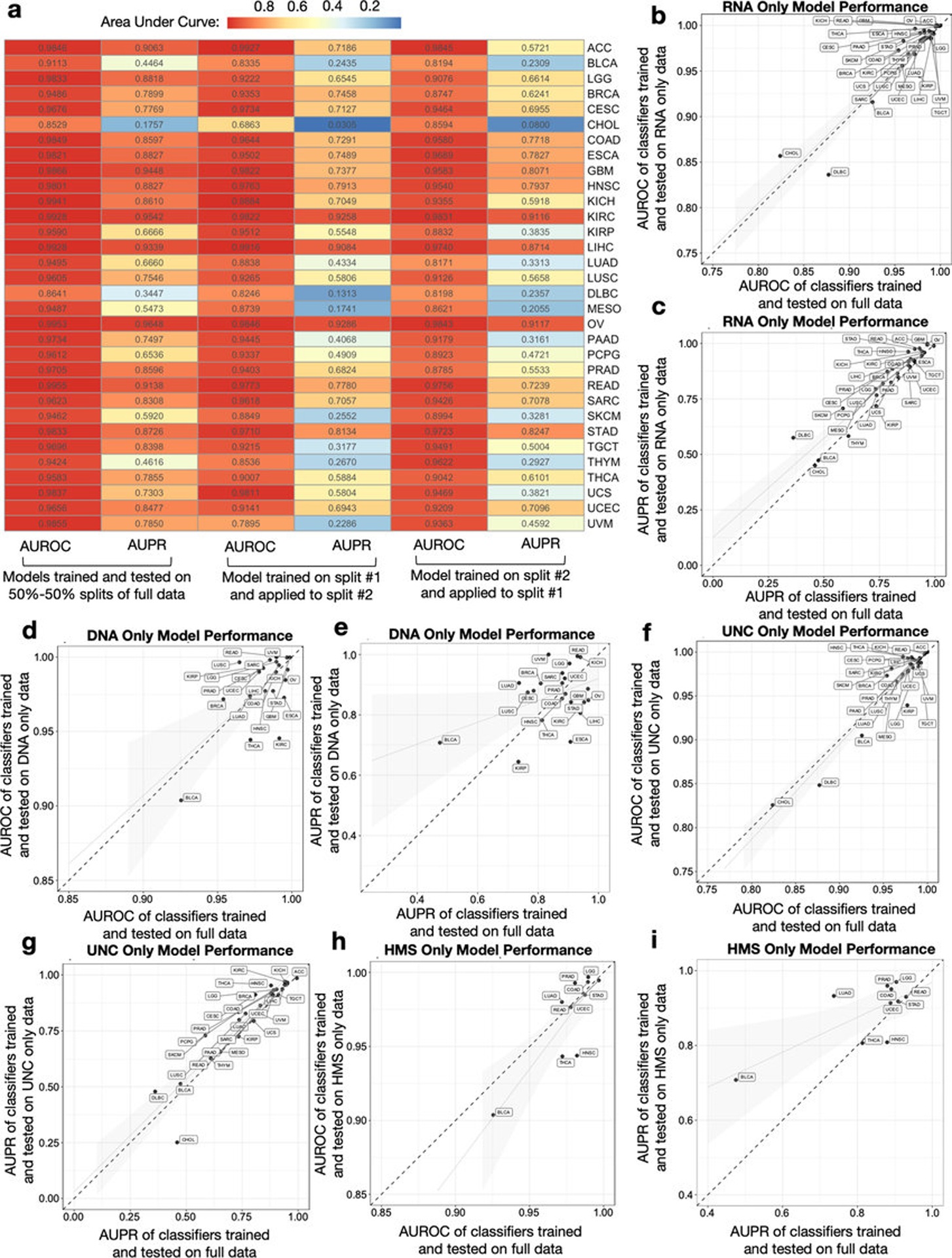
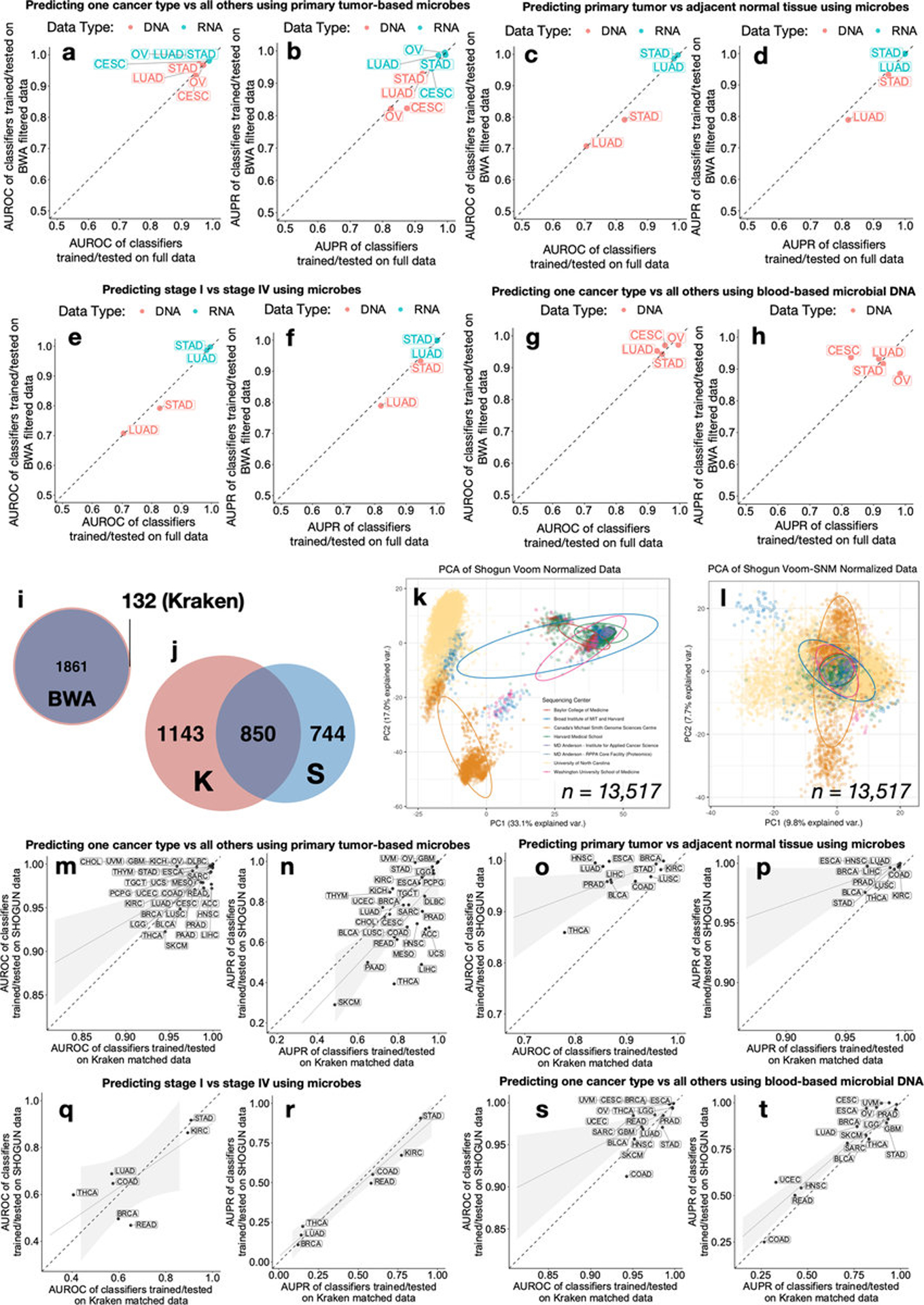
Comment in
-
AI finds microbial signatures in tumours and blood across cancer types.Nature. 2020 Mar;579(7800):502-503. doi: 10.1038/d41586-020-00637-w. Nature. 2020. PMID: 32161344 No abstract available.
-
Microbial Diagnostics for Cancer: A Step Forward but Not Prime Time Yet.Cancer Cell. 2020 May 11;37(5):625-627. doi: 10.1016/j.ccell.2020.04.010. Cancer Cell. 2020. PMID: 32396856
-
Microbial DNA signature in plasma enables cancer diagnosis.Nat Rev Clin Oncol. 2020 Aug;17(8):453-454. doi: 10.1038/s41571-020-0391-1. Nat Rev Clin Oncol. 2020. PMID: 32424197 No abstract available.
-
Microbiome-Derived Liquid Biopsy: New Hope for Cancer Screening?Clin Chem. 2021 Mar 1;67(3):463-465. doi: 10.1093/clinchem/hvaa240. Clin Chem. 2021. PMID: 33277898 Free PMC article. No abstract available.
-
Caution regarding the specificities of pan-cancer microbial structure.Microb Genom. 2023 Aug;9(8):mgen001088. doi: 10.1099/mgen.0.001088. Microb Genom. 2023. PMID: 37555750 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
