

MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis
- PMID: 32220583
- PMCID: PMC7438259
- DOI: 10.1016/j.jhep.2020.03.023
MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis
Erratum in
-
Erratum to: "MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcoholic-associated fatty liver and steatohepatitis (J Hepatol 2020; 73: 616-627)".J Hepatol. 2021 Apr;74(4):1002. doi: 10.1016/j.jhep.2021.01.006. Epub 2021 Jan 30. J Hepatol. 2021. PMID: 33526291 No abstract available.
Abstract
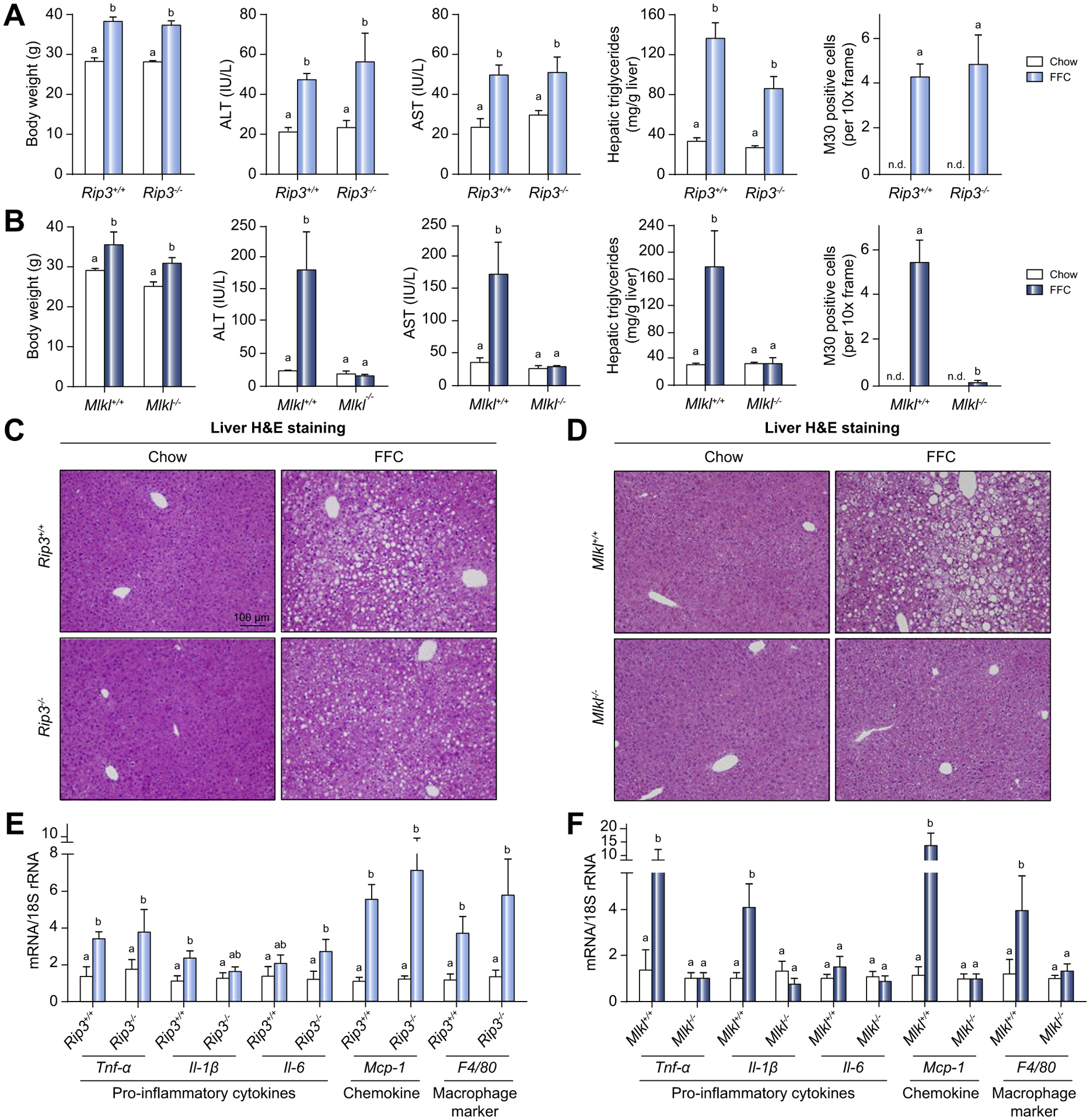
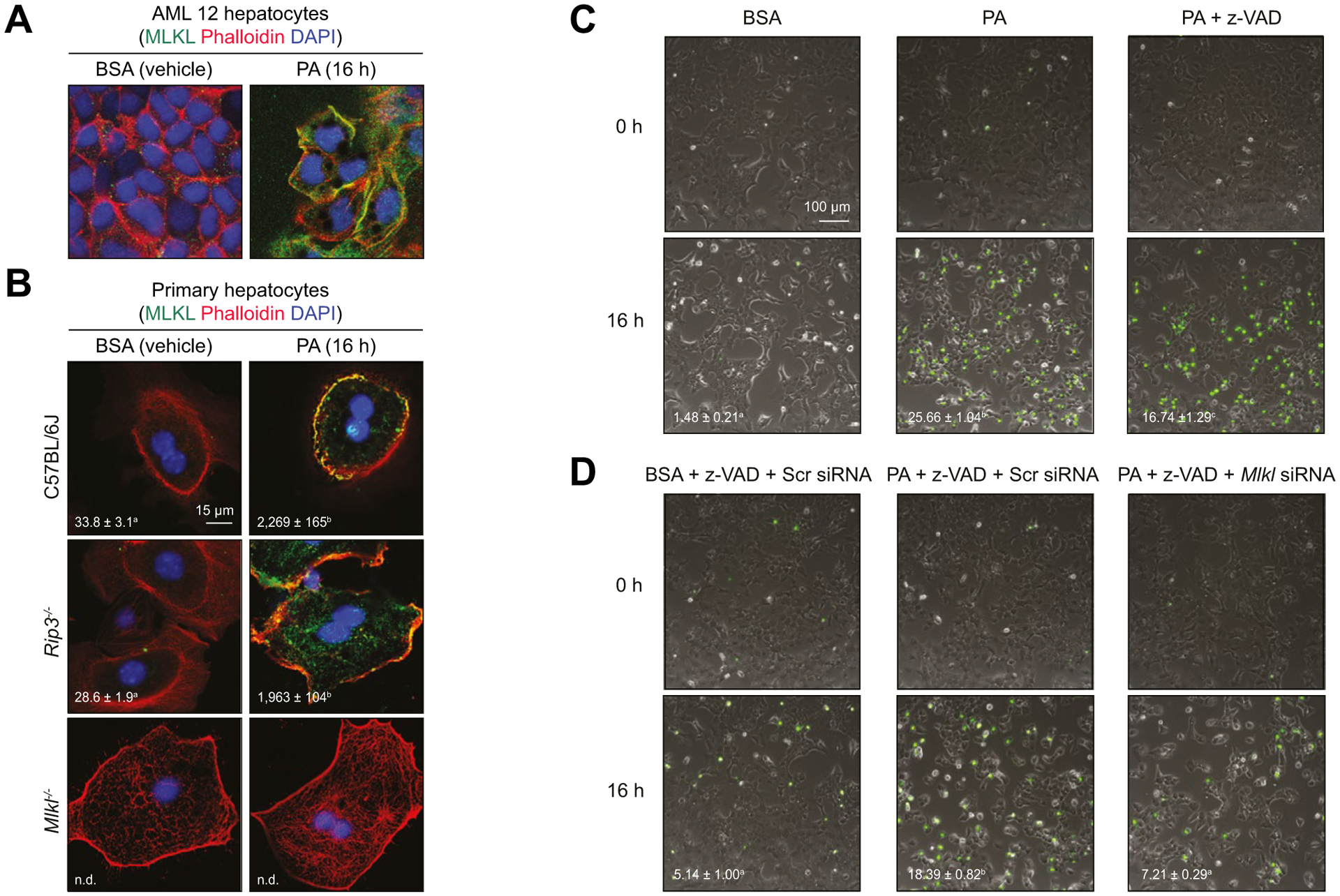
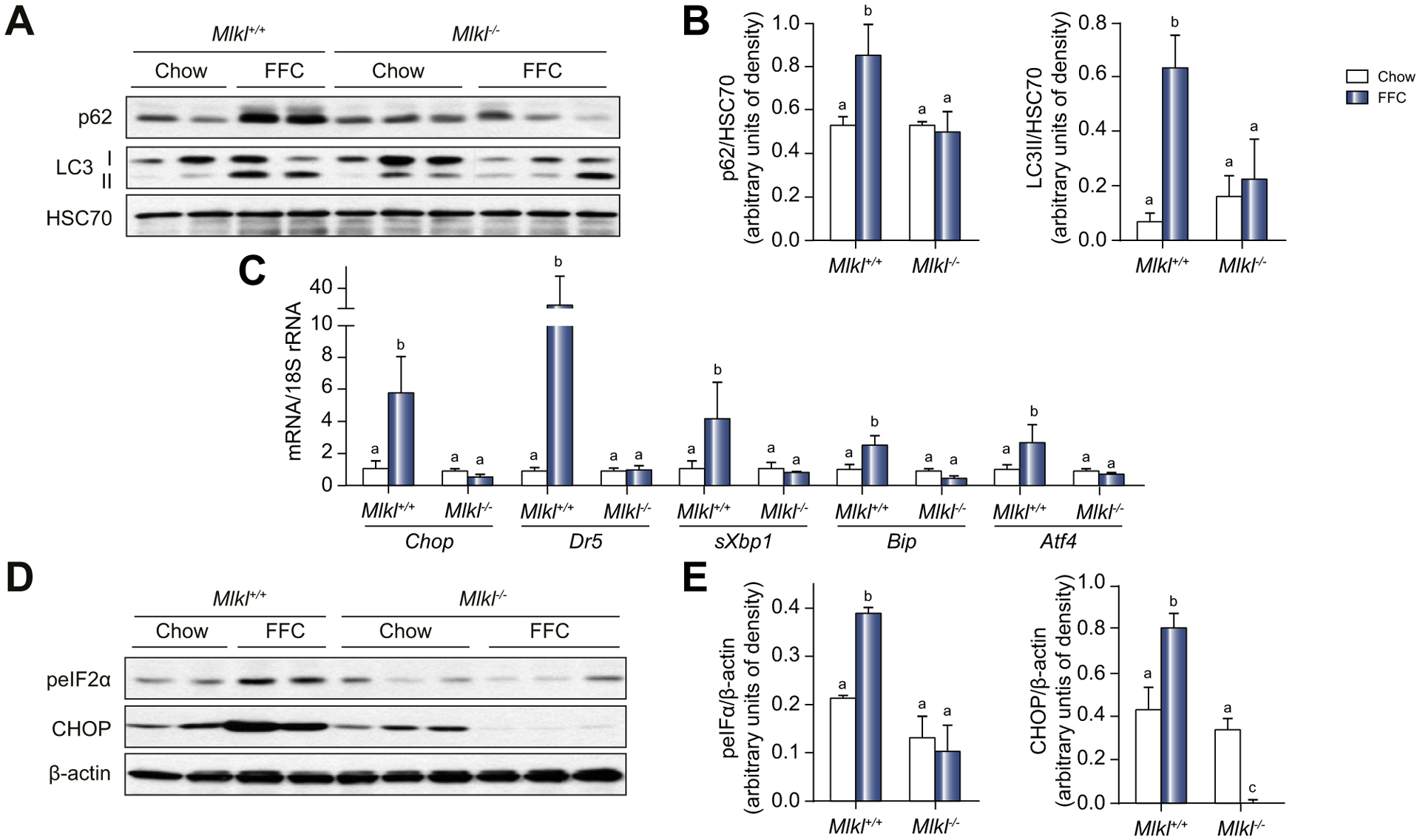
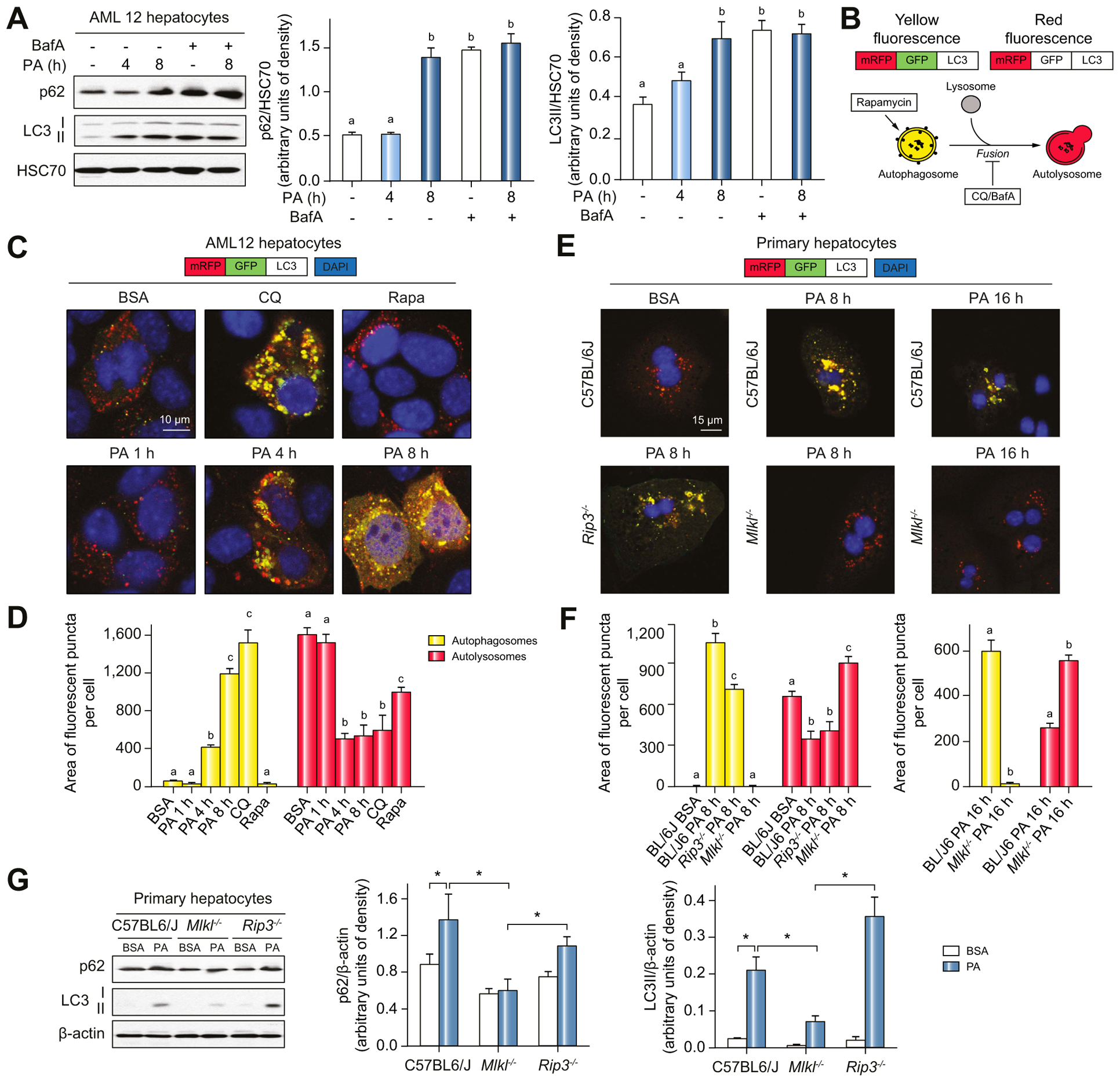
Background & aims: Autophagy maintains cellular homeostasis and plays a critical role in the development of non-alcoholic fatty liver and steatohepatitis. The pseudokinase mixed lineage kinase domain-like (MLKL) is a key downstream effector of receptor interacting protein kinase 3 (RIP3) in the necroptotic pathway of programmed cell death. However, recent data reveal that MLKL also regulates autophagy. Herein, we tested the hypothesis that MLKL contributes to the progression of Western diet-induced liver injury in mice by regulating autophagy.
Methods: Rip3+/+, Rip3-/-, Mlkl+/+ and Mlkl-/- mice were fed a Western diet (FFC diet, high in fat, fructose and cholesterol) or chow for 12 weeks. AML12 and primary mouse hepatocytes were exposed to palmitic acid (PA).
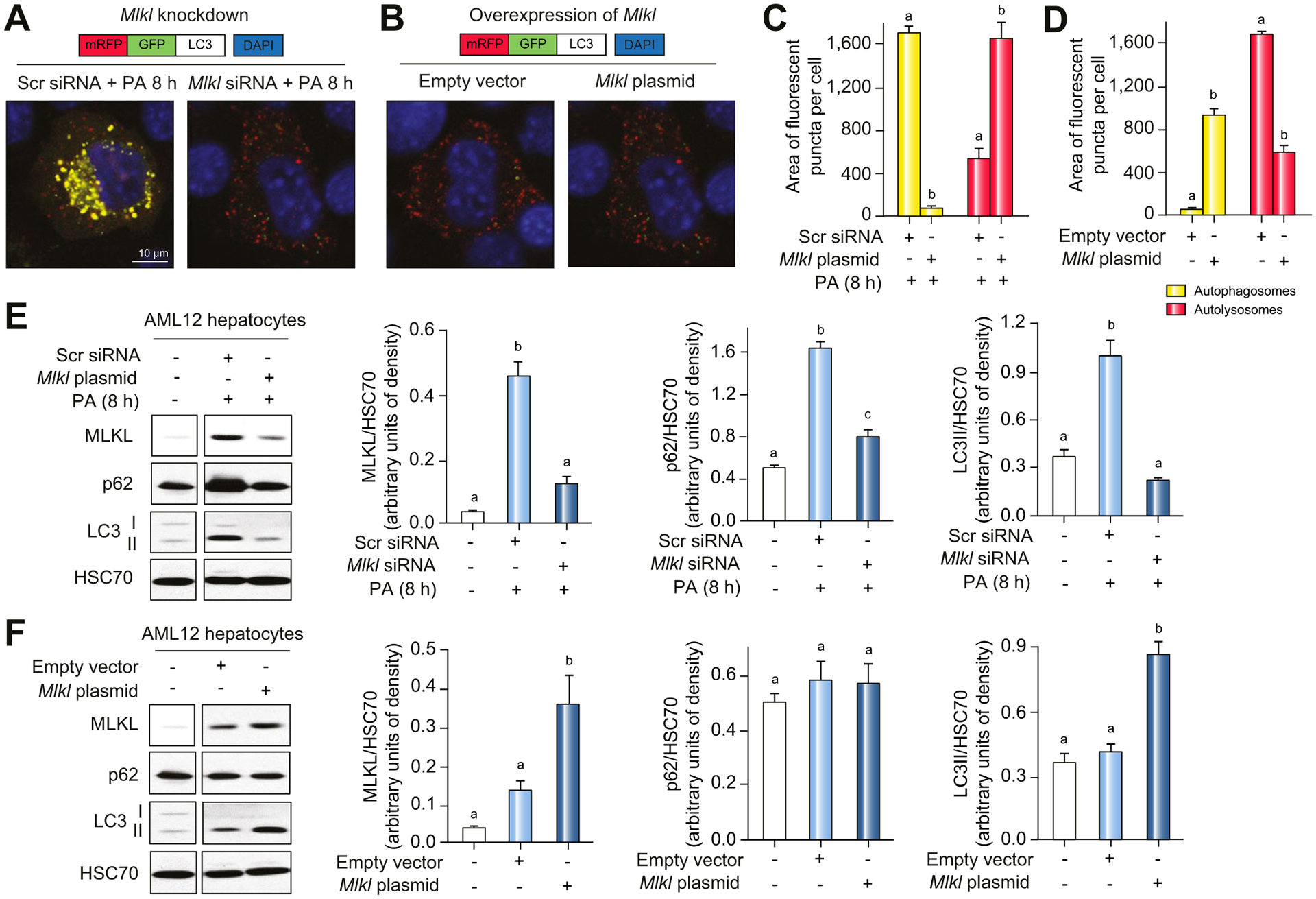
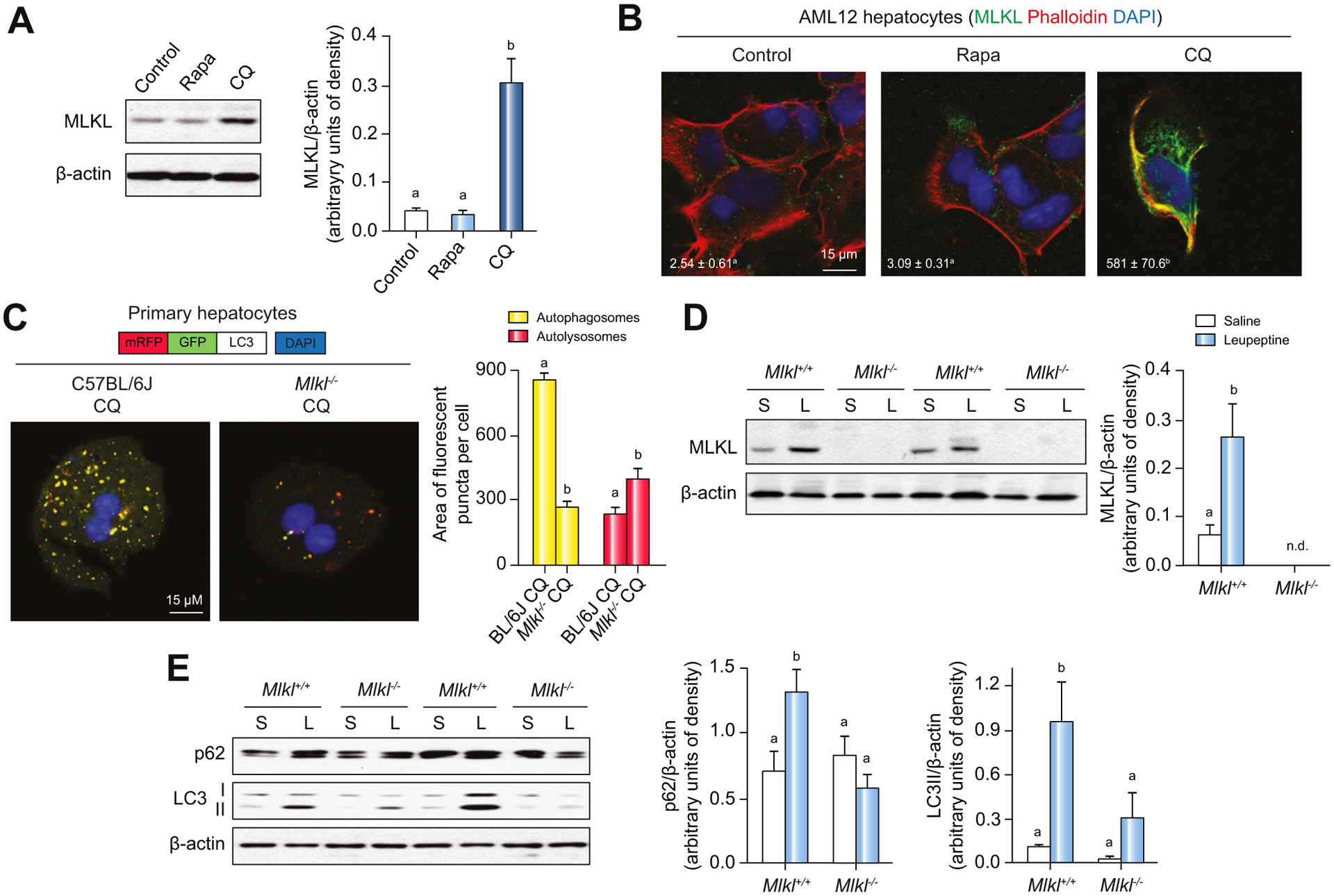
Results: The FFC diet increased expression, phosphorylation and oligomerization of MLKL in the liver. Mlkl, but not Rip3, deficiency protected mice from FFC diet-induced liver injury. The FFC diet also induced accumulation of p62 and LC3-II, as well as markers of endoplasmic reticulum stress, in Mlkl+/+ but not Mlkl-/- mice. Mlkl deficiency in mice also prevented the inhibition of autophagy by a protease inhibitor, leupeptin. Using an mRFP-GFP-LC3 reporter in cultured hepatocytes revealed that PA blocked the fusion of autophagosomes with lysosomes. PA triggered MLKL expression and translocation, first to autophagosomes and then to the plasma membrane, independently of Rip3. Mlkl, but not Rip3, deficiency prevented inhibition of autophagy in PA-treated hepatocytes. Overexpression of Mlkl blocked autophagy independently of PA. Additionally, pharmacologic inhibition of autophagy induced MLKL expression and translocation to the plasma membrane in hepatocytes.
Conclusions: Taken together, these data indicate that MLKL-dependent, but RIP3-independent, signaling contributes to FFC diet-induced liver injury by inhibiting autophagy.
Lay summary: Autophagy is a regulated process that maintains cellular homeostasis. Impaired autophagy contributes to cell injury and death, thus playing a critical role in the pathogenesis of a number of diseases, including non-alcohol-associated fatty liver and steatohepatitis. Herein, we show that Mlkl-dependent, but Rip3-independent, signaling contributed to diet-induced liver injury and inflammatory responses by inhibiting autophagy. These data identify a novel co-regulatory mechanism between necroptotic and autophagic signaling pathways in non-alcoholic fatty liver disease.
Keywords: Autophagic flux; MLKL; NAFLD; NASH; Necroptosis; RIPK3.
Copyright © 2020 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare no conflicts of interest that pertain to this work. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures


References
-
- Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol 2013;59:583–594. - PubMed
-
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008;133:693–703. - PubMed
-
- Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 2015;15:362–374. - PubMed
-
- Hatting M, Zhao G, Schumacher F, Sellge G, Al Masaoudi M, Gabetaler N, et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology (Baltimore, Md) 2013;57:2189–2201. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 AA026938/AA/NIAAA NIH HHS/United States
- R01 AA023722/AA/NIAAA NIH HHS/United States
- R21 AA020941/AA/NIAAA NIH HHS/United States
- R00 AA026648/AA/NIAAA NIH HHS/United States
- R01 AA027456/AA/NIAAA NIH HHS/United States
- R01 DK113196/DK/NIDDK NIH HHS/United States
- P50 AA024333/AA/NIAAA NIH HHS/United States
- U01 AA020821/AA/NIAAA NIH HHS/United States
- R01 GM119174/GM/NIGMS NIH HHS/United States
- K99 AA026648/AA/NIAAA NIH HHS/United States
- R21 AR071046/AR/NIAMS NIH HHS/United States
- P30 DK097948/DK/NIDDK NIH HHS/United States
- U01 AA021890/AA/NIAAA NIH HHS/United States
- U01 AA026976/AA/NIAAA NIH HHS/United States
- S10 RR026820/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
