

Constitutive Activation of RAS/MAPK Pathway Cooperates with Trisomy 21 and Is Therapeutically Exploitable in Down Syndrome B-cell Leukemia
- PMID: 32220889
- PMCID: PMC7334063
- DOI: 10.1158/1078-0432.CCR-19-3519
Constitutive Activation of RAS/MAPK Pathway Cooperates with Trisomy 21 and Is Therapeutically Exploitable in Down Syndrome B-cell Leukemia
Abstract
Purpose: Children with Down syndrome (constitutive trisomy 21) that develop acute lymphoblastic leukemia (DS-ALL) have a 3-fold increased likelihood of treatment-related mortality coupled with a higher cumulative incidence of relapse, compared with other children with B-cell acute lymphoblastic leukemia (B-ALL). This highlights the lack of suitable treatment for Down syndrome children with B-ALL.
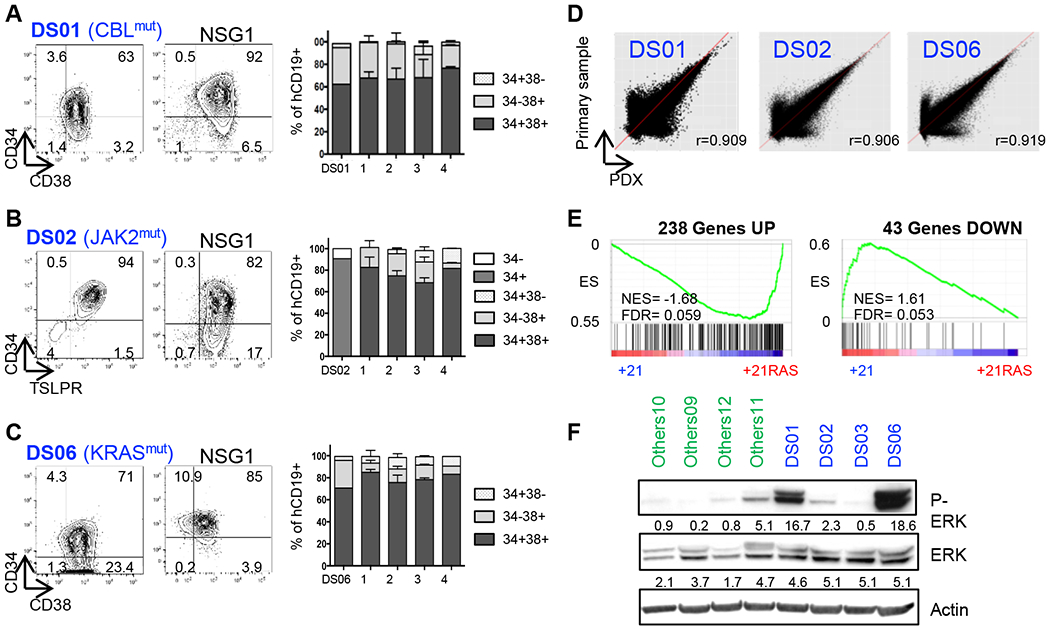
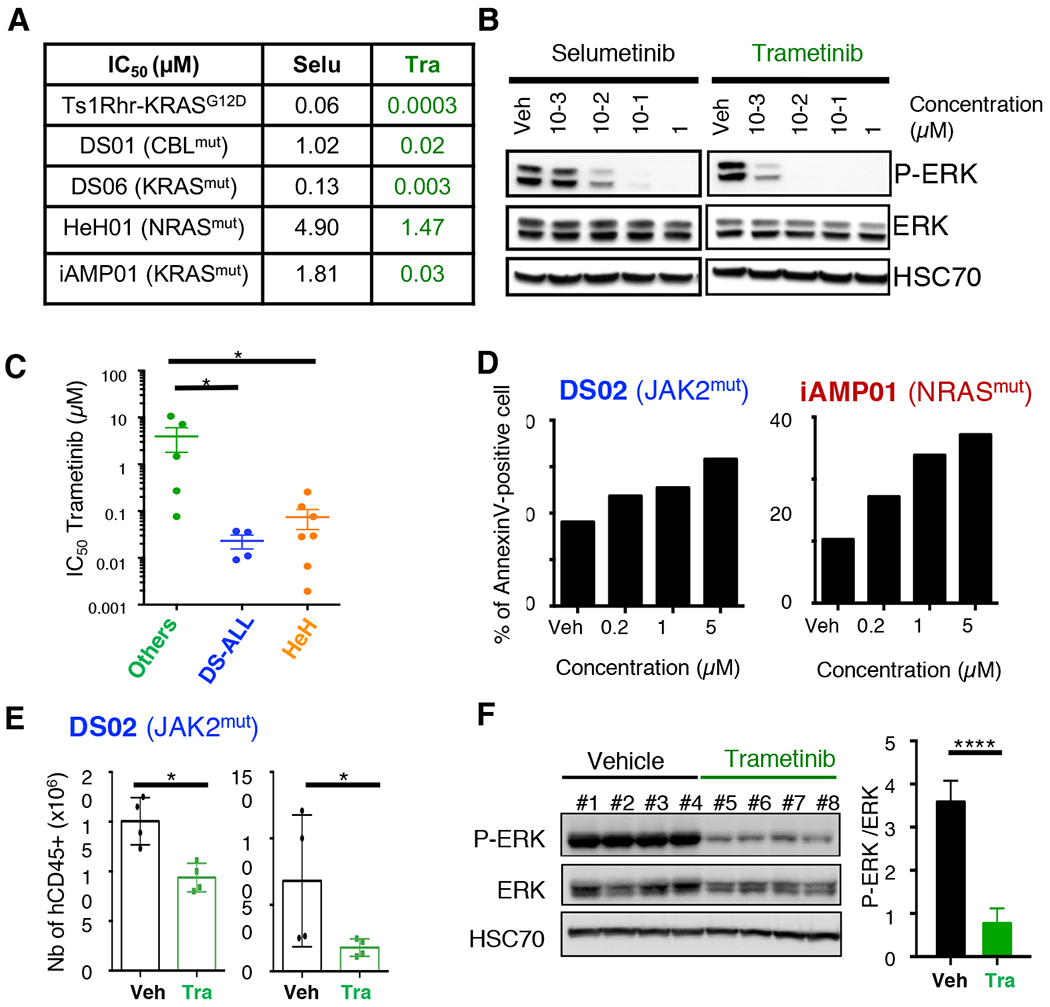
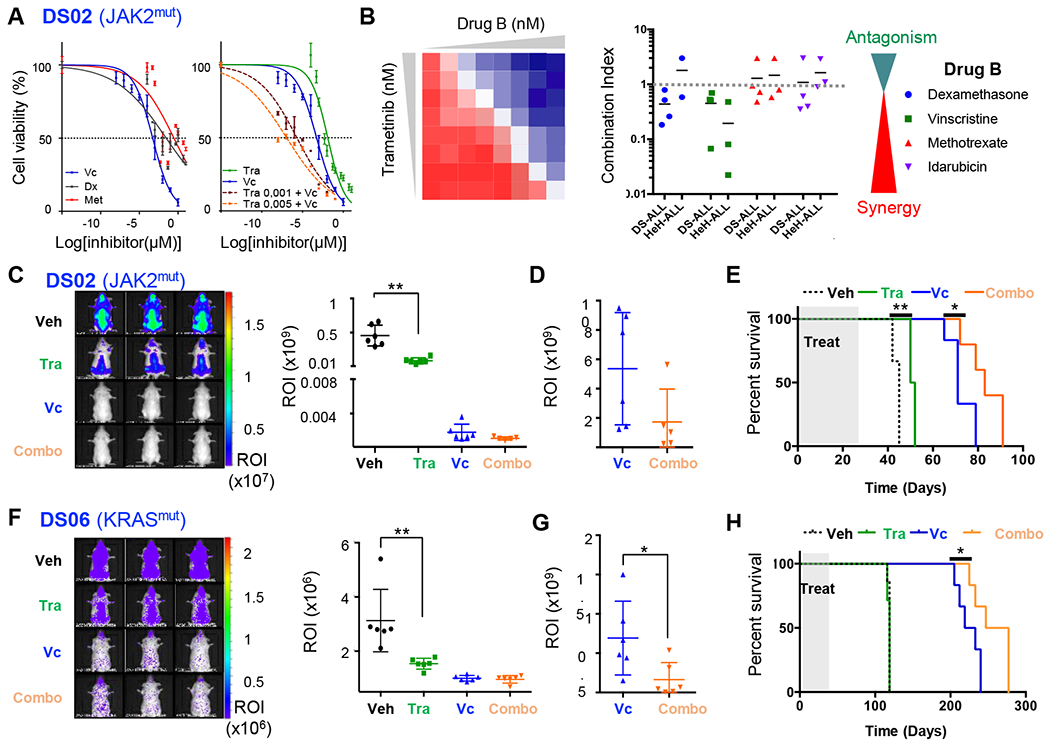
Experimental design: To facilitate the translation of new therapeutic agents into clinical trials, we built the first preclinical cohort of patient-derived xenograft (PDX) models of DS-ALL, comprehensively characterized at the genetic and transcriptomic levels, and have proven its suitability for preclinical studies by assessing the efficacy of drug combination between the MEK inhibitor trametinib and conventional chemotherapy agents.
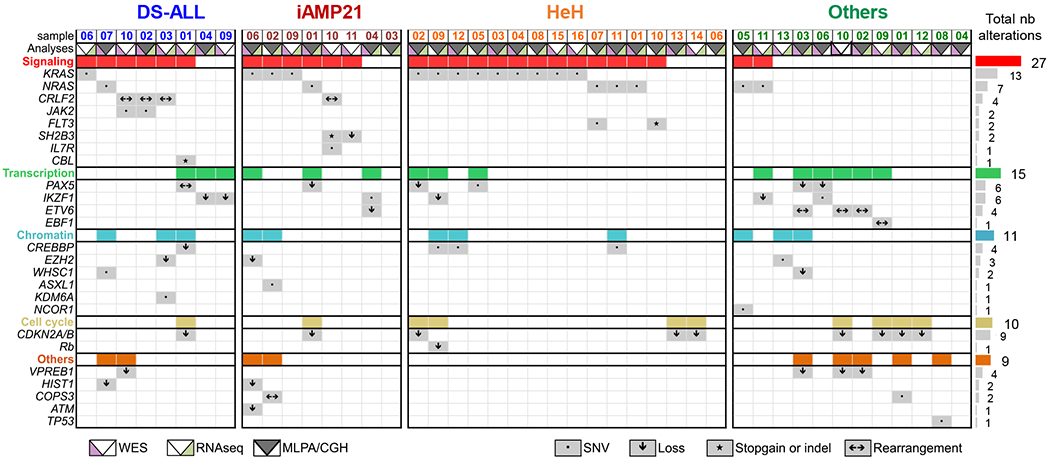
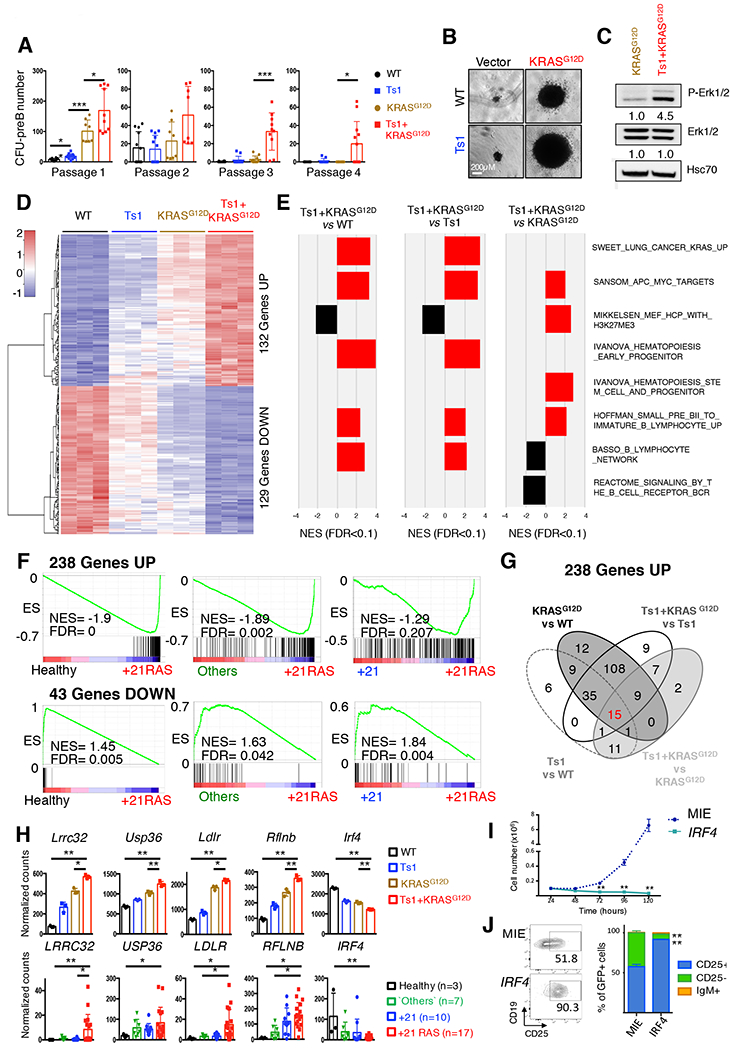
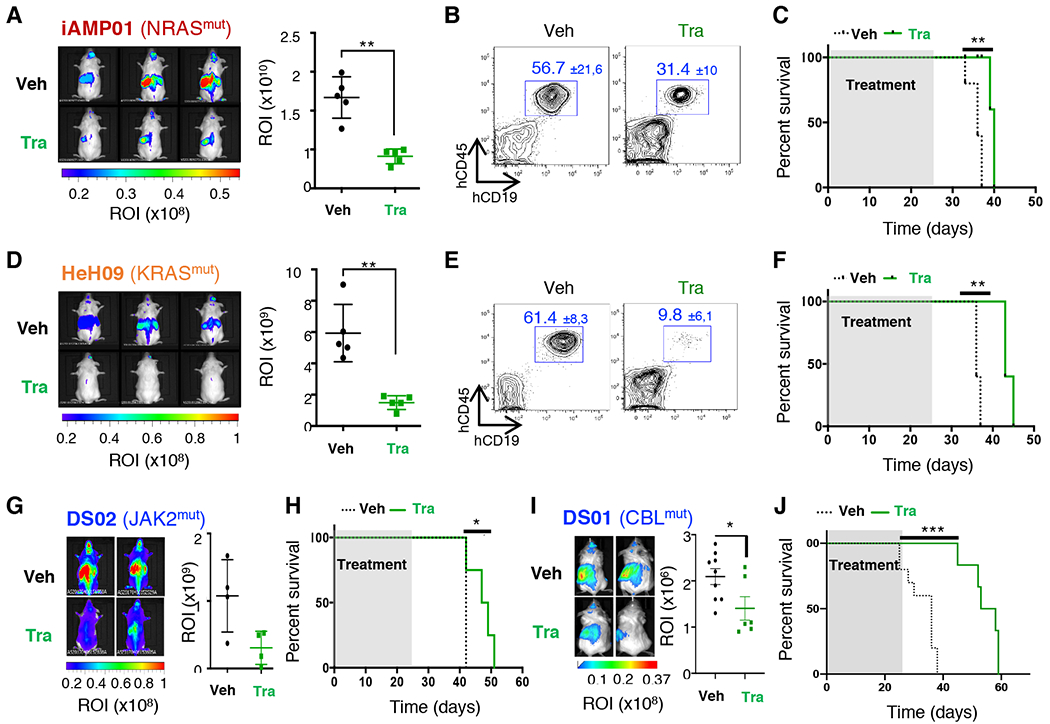
Results: Whole-exome and RNA-sequencing experiments revealed a high incidence of somatic alterations leading to RAS/MAPK pathway activation in our cohort of DS-ALL, as well as in other pediatric B-ALL presenting somatic gain of the chromosome 21 (B-ALL+21). In murine and human B-cell precursors, activated KRASG12D functionally cooperates with trisomy 21 to deregulate transcriptional networks that promote increased proliferation and self renewal, as well as B-cell differentiation blockade. Moreover, we revealed that inhibition of RAS/MAPK pathway activation using the MEK1/2 inhibitor trametinib decreased leukemia burden in several PDX models of B-ALL+21, and enhanced survival of DS-ALL PDX in combination with conventional chemotherapy agents such as vincristine.
Conclusions: Altogether, using novel and suitable PDX models, this study indicates that RAS/MAPK pathway inhibition represents a promising strategy to improve the outcome of Down syndrome children with B-cell precursor leukemia.
©2020 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest
JDC receives research support from Forma Therapeutics and Scholar Rock, is a consultant for Sierra Oncology, and the Scientific Advisor of the MPN Research Foundation. The other authors declare no conflict of interest.
Figures
References
-
- Hasle H, Friedman JM, Olsen JH, Rasmussen SA. Low risk of solid tumors in persons with Down syndrome. Genet Med. 2016;18(11):1151–7. - PubMed
-
- Ceppi F, Stephens D, den Hollander BS, Krueger J, Whitlock J, Sung L, et al. Clinical presentation and risk factors of serious infections in children with Down syndrome treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 2016;63(11):1949–53. - PubMed
-
- Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, Michalova K, et al. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111(3):1575–83. - PubMed
-
- Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688–98. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
