

Mapping RNA splicing variations in clinically accessible and nonaccessible tissues to facilitate Mendelian disease diagnosis using RNA-seq
- PMID: 32225167
- PMCID: PMC7335339
- DOI: 10.1038/s41436-020-0780-y
Mapping RNA splicing variations in clinically accessible and nonaccessible tissues to facilitate Mendelian disease diagnosis using RNA-seq
Abstract
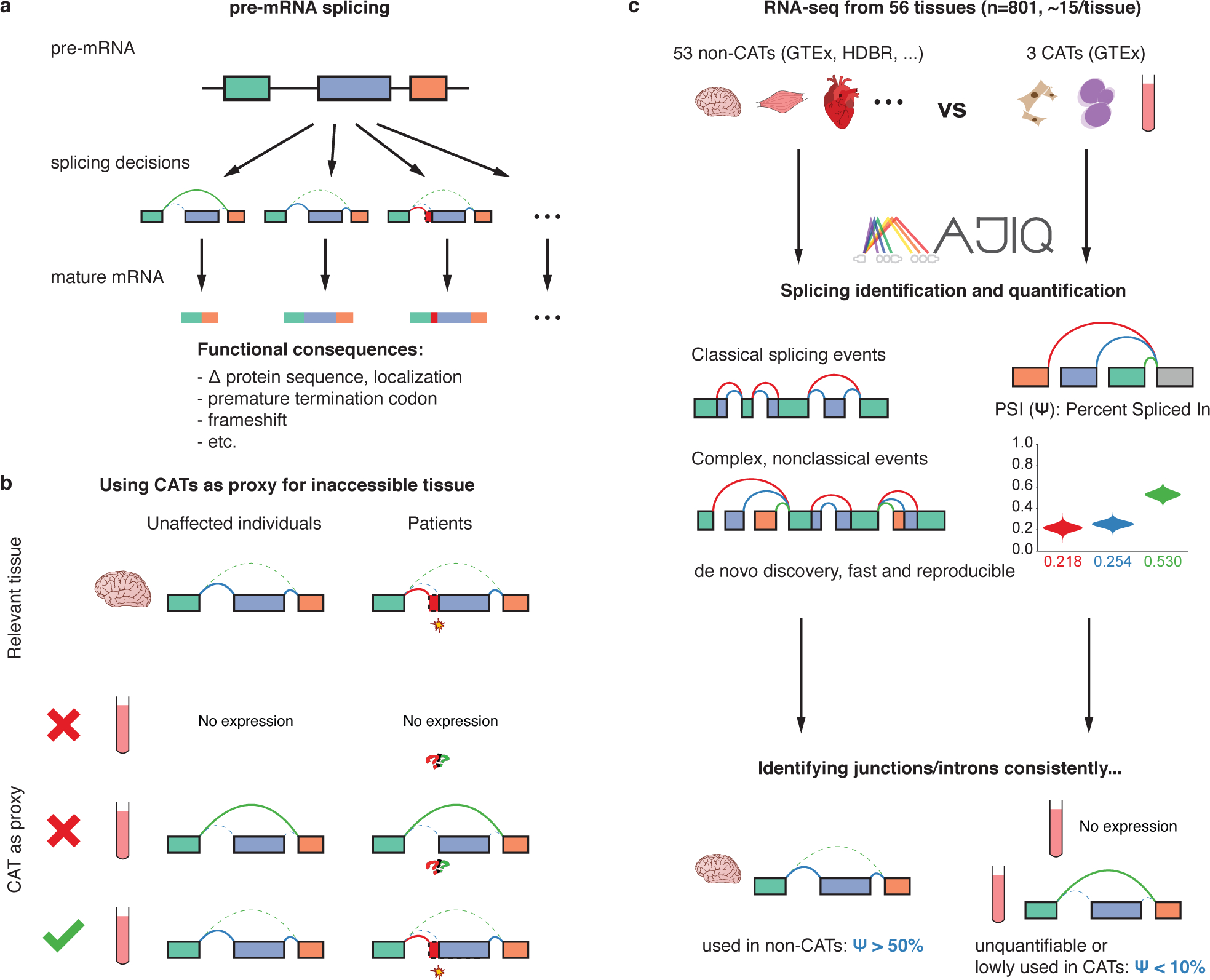
Purpose: RNA-seq is a promising approach to improve diagnoses by detecting pathogenic aberrations in RNA splicing that are missed by DNA sequencing. RNA-seq is typically performed on clinically accessible tissues (CATs) from blood and skin. RNA tissue specificity makes it difficult to identify aberrations in relevant but nonaccessible tissues (non-CATs). We determined how RNA-seq from CATs represent splicing in and across genes and non-CATs.
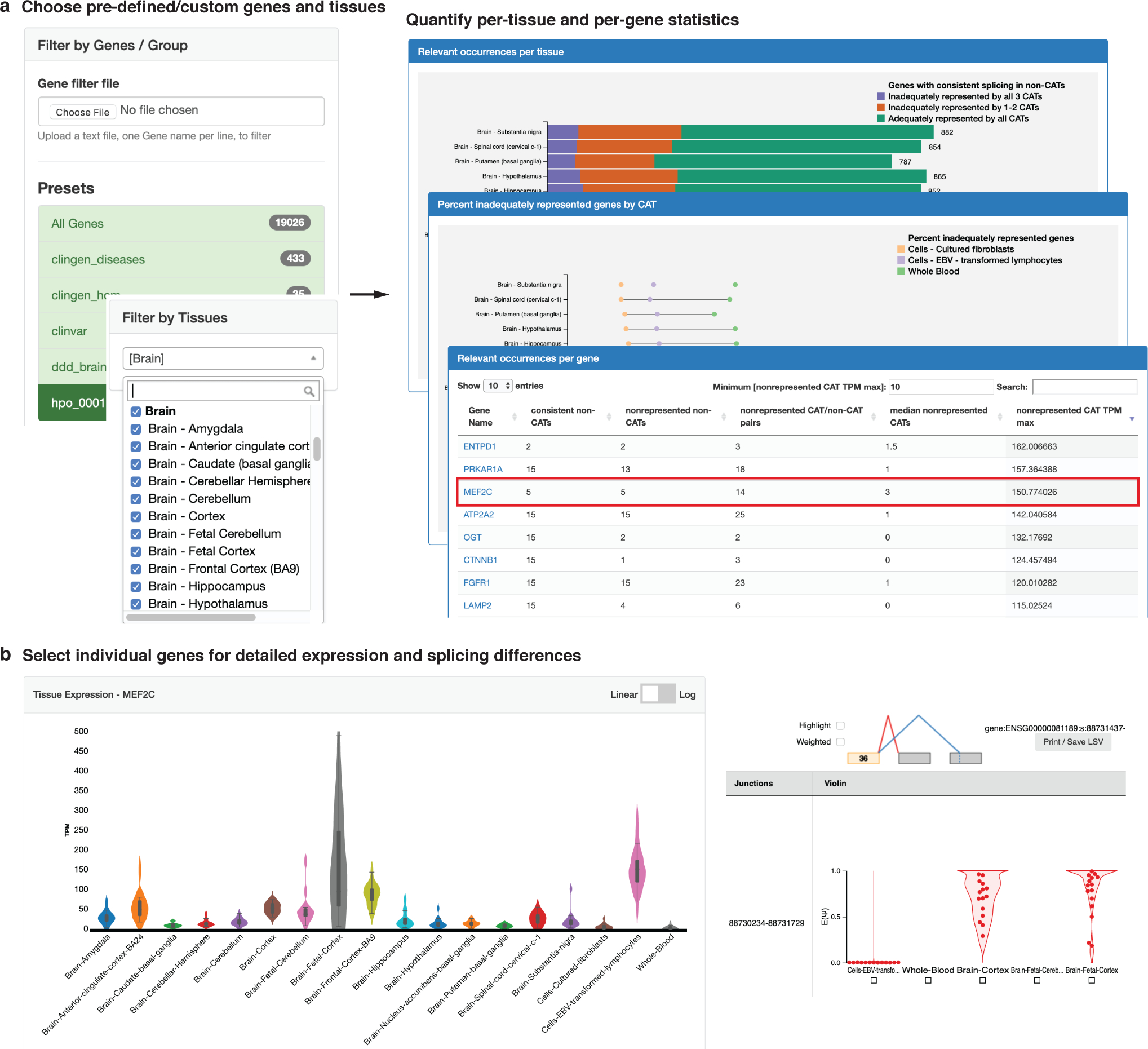
Methods: We quantified RNA splicing in 801 RNA-seq samples from 56 different adult and fetal tissues from Genotype-Tissue Expression Project (GTEx) and ArrayExpress. We identified genes and splicing events in each non-CAT and determined when RNA-seq in each CAT would inadequately represent them. We developed an online resource, MAJIQ-CAT, for exploring our analysis for specific genes and tissues.
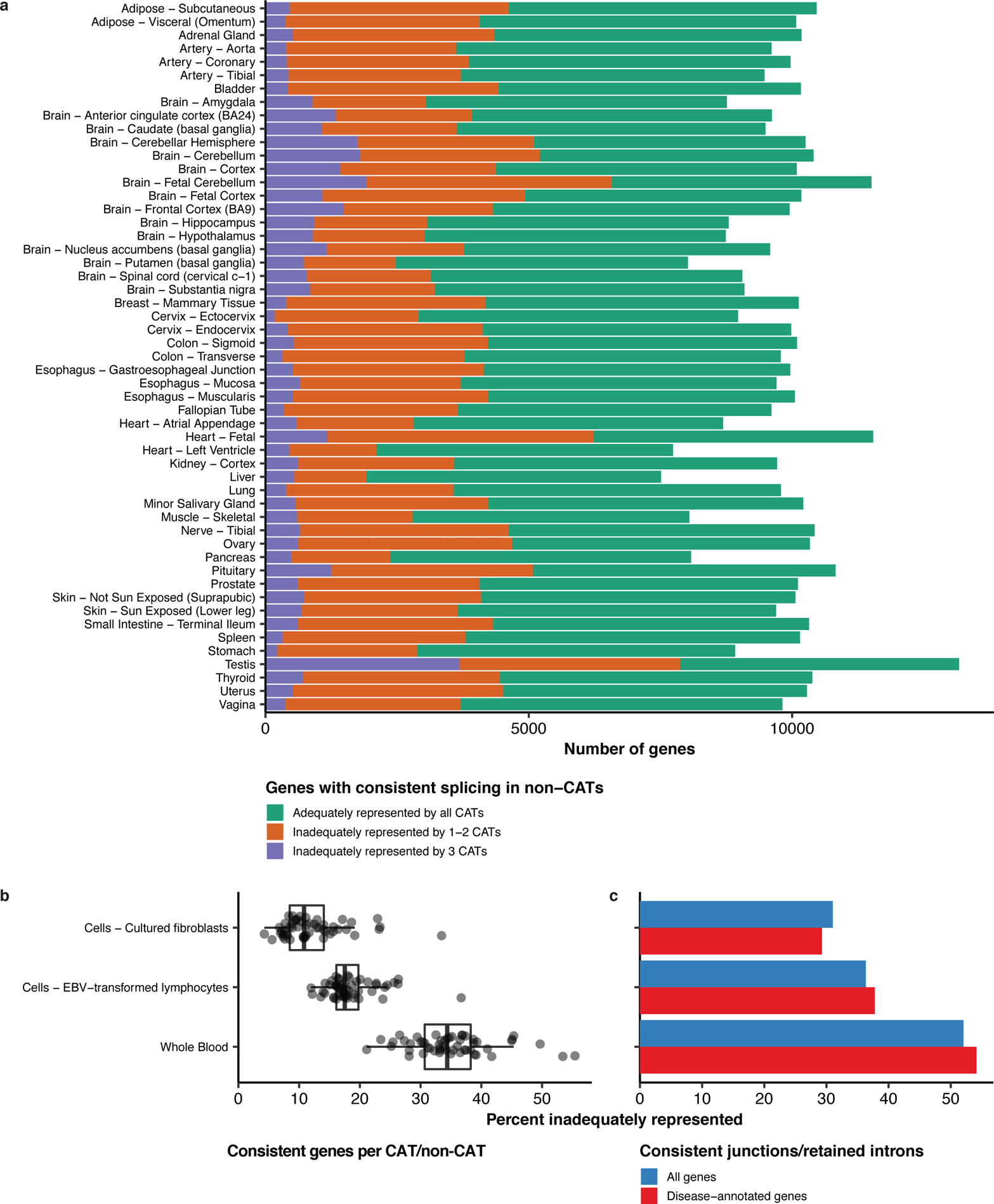
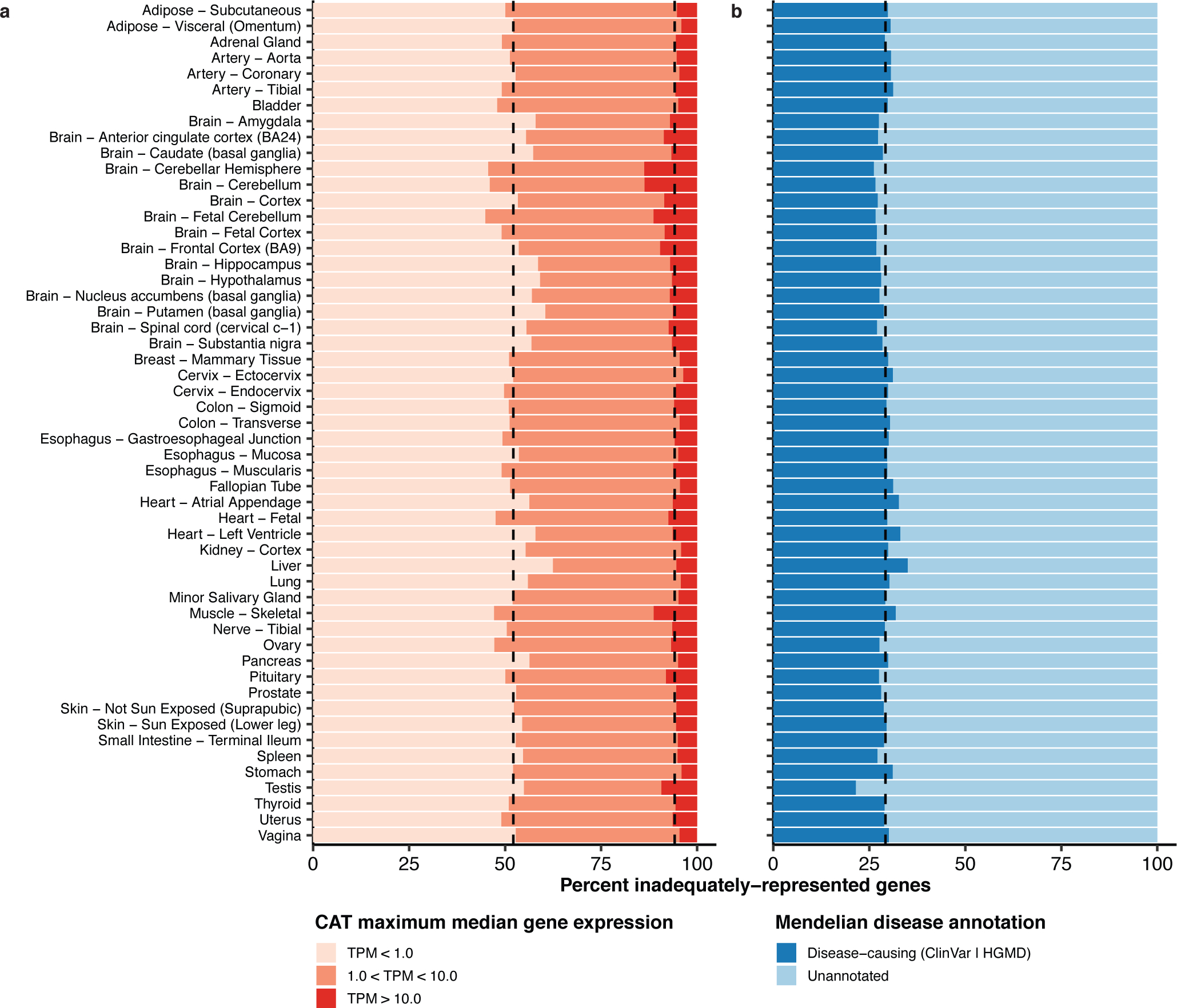
Results: In non-CATs, 40.2% of genes have splicing that is inadequately represented by at least one CAT; 6.3% of genes have splicing inadequately represented by all CATs. A majority (52.1%) of inadequately represented genes are lowly expressed in CATs (transcripts per million (TPM) < 1), but 5.8% are inadequately represented despite being well expressed (TPM > 10).
Conclusion: Many splicing events in non-CATs are inadequately evaluated using RNA-seq from CATs. MAJIQ-CAT allows users to explore which accessible tissues, if any, best represent splicing in genes and tissues of interest.
Keywords: RNA-seq; alternative splicing; clinical genetics; diagnostic markers; medical genetics.
Figures
References
-
- Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med Off J Am Coll Med Genet. 2015;17(7):578–586. doi:10.1038/gim.2014.154 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
