

Advances in Knowledge of Candidate Genes Acting at the Beta-Cell Level in the Pathogenesis of T1DM
- PMID: 32226409
- PMCID: PMC7080653
- DOI: 10.3389/fendo.2020.00119
Advances in Knowledge of Candidate Genes Acting at the Beta-Cell Level in the Pathogenesis of T1DM
Abstract
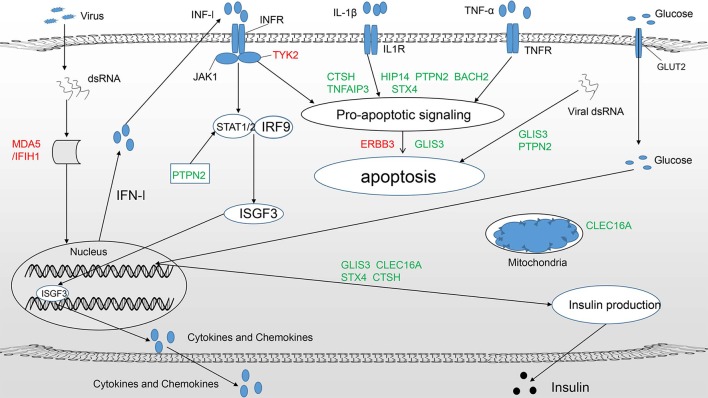
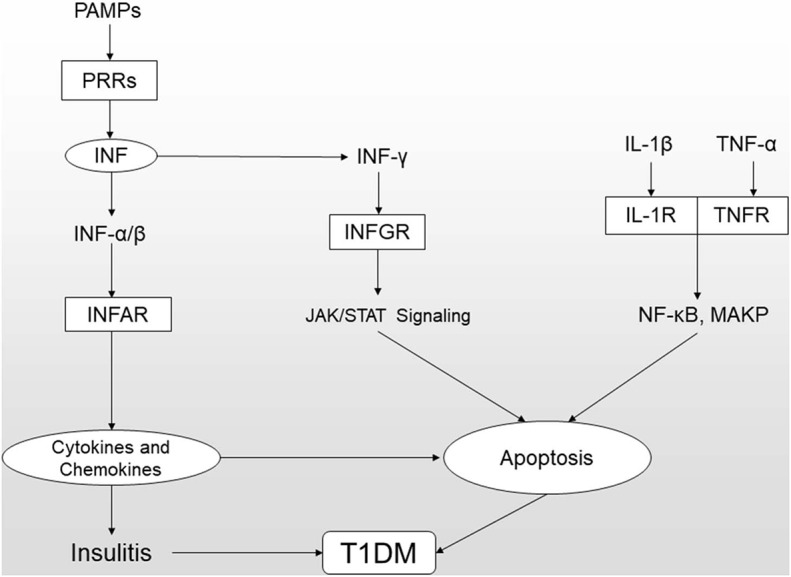
T1DM (type 1 diabetes mellitus), which results from the irreversible elimination of beta-cells mediated by autoreactive T cells, is defined as an autoimmune disease. It is widely accepted that T1DM is caused by a combination of genetic and environmental factors, but the precise underlying molecular mechanisms are still unknown. To date, more than 50 genetic risk regions contributing to the pathogenesis of T1DM have been identified by GWAS (genome-wide association studies). Notably, more than 60% of the identified candidate genes are expressed in islets and beta-cells, which makes it plausible that these genes act at the beta-cell level and play a key role in the pathogenesis of T1DM. In this review, we focus on the current status of candidate genes that act at the beta-cell level by regulating the innate immune response and antiviral activity, affecting susceptibility to proapoptotic stimuli and influencing the pancreatic beta-cell phenotype.
Keywords: GWAS; T1DM; apoptosis; beta-cell phenotype; candidate gene; innate immunity; pancreatic beta-cell.
Copyright © 2020 Pang, Luo, Huang, Xia, Xie and Zhou.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical