

Nuclear receptor ERRα contributes to castration-resistant growth of prostate cancer via its regulation of intratumoral androgen biosynthesis
- PMID: 32226548
- PMCID: PMC7086365
- DOI: 10.7150/thno.35589
Nuclear receptor ERRα contributes to castration-resistant growth of prostate cancer via its regulation of intratumoral androgen biosynthesis
Abstract
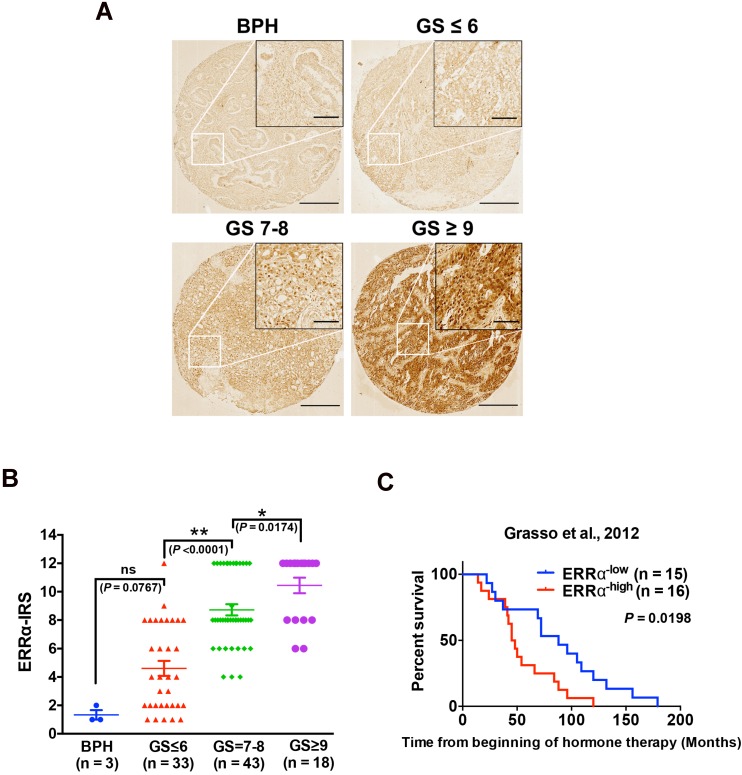
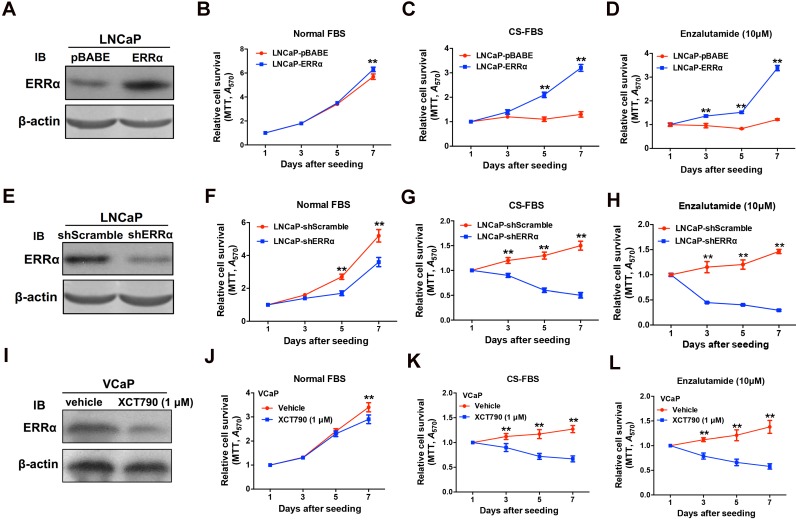
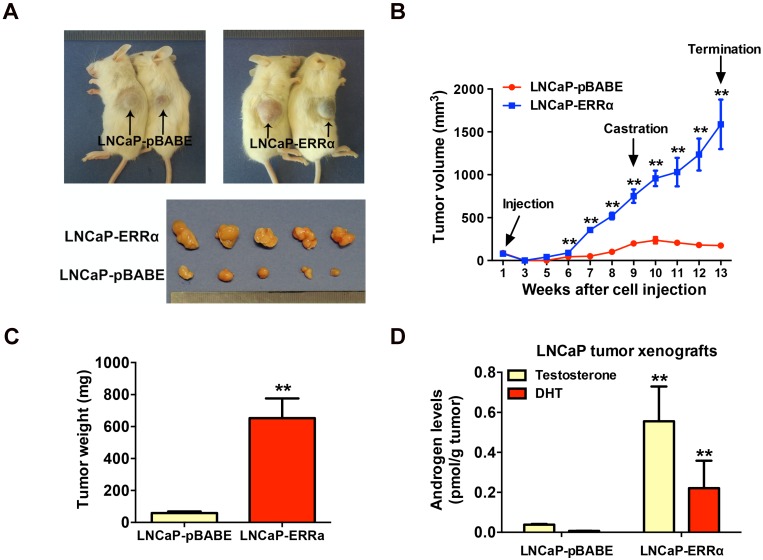
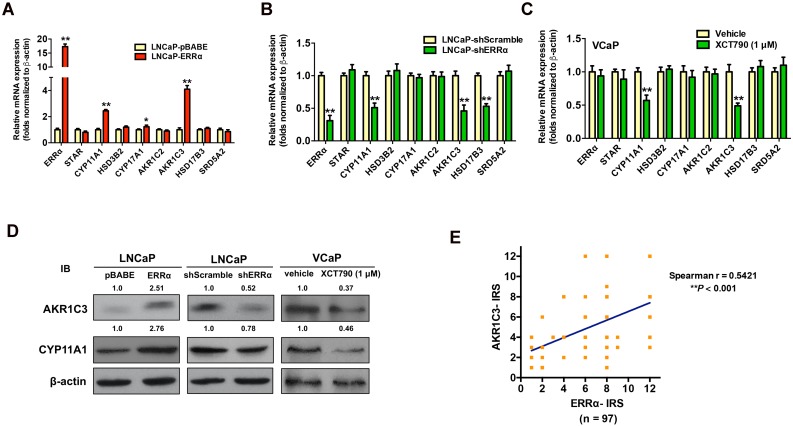
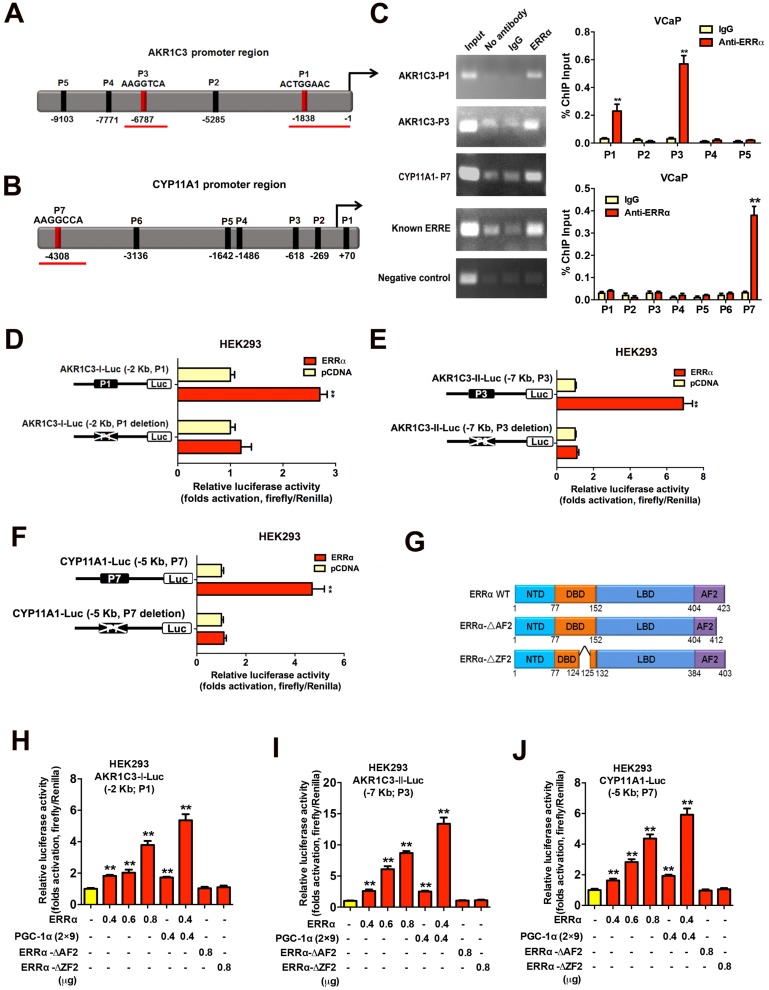
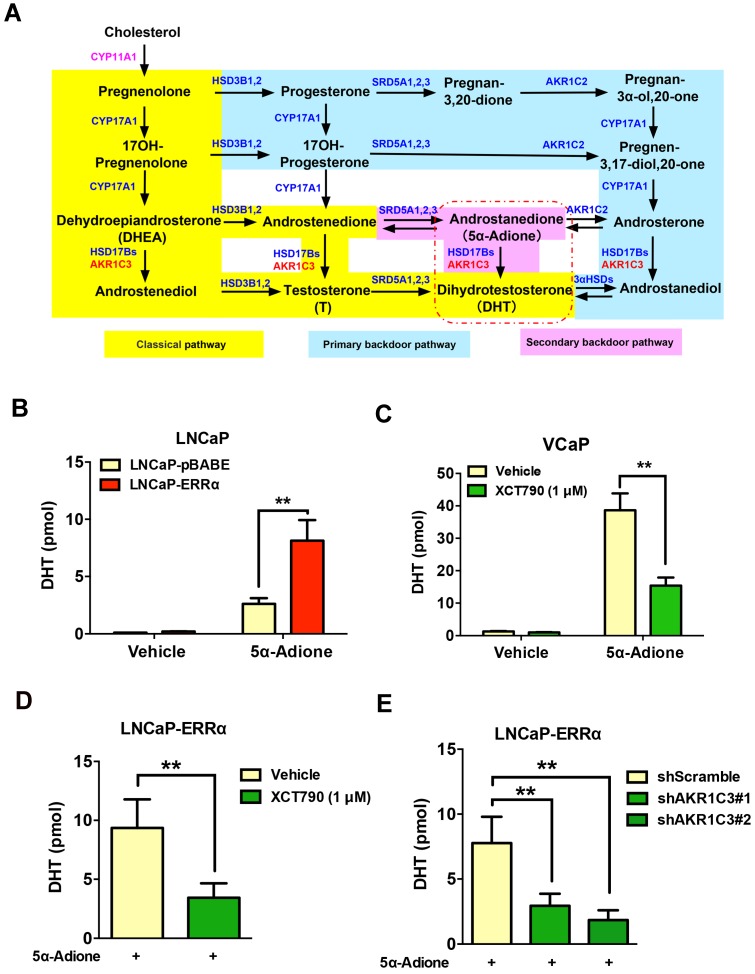
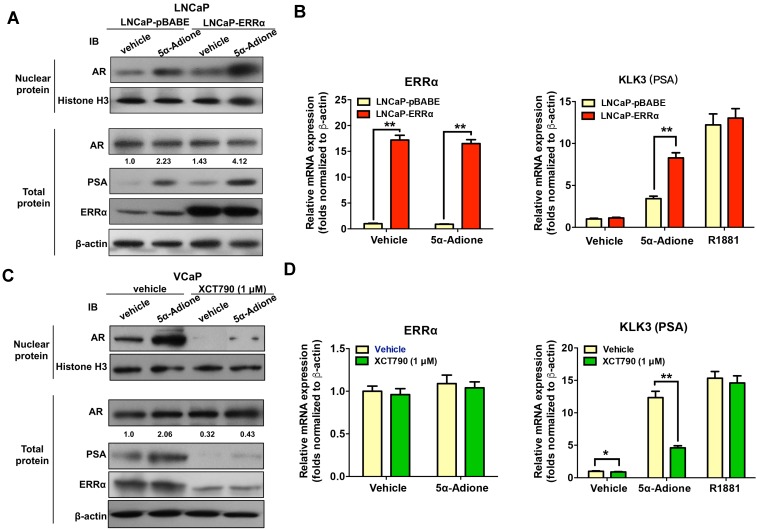
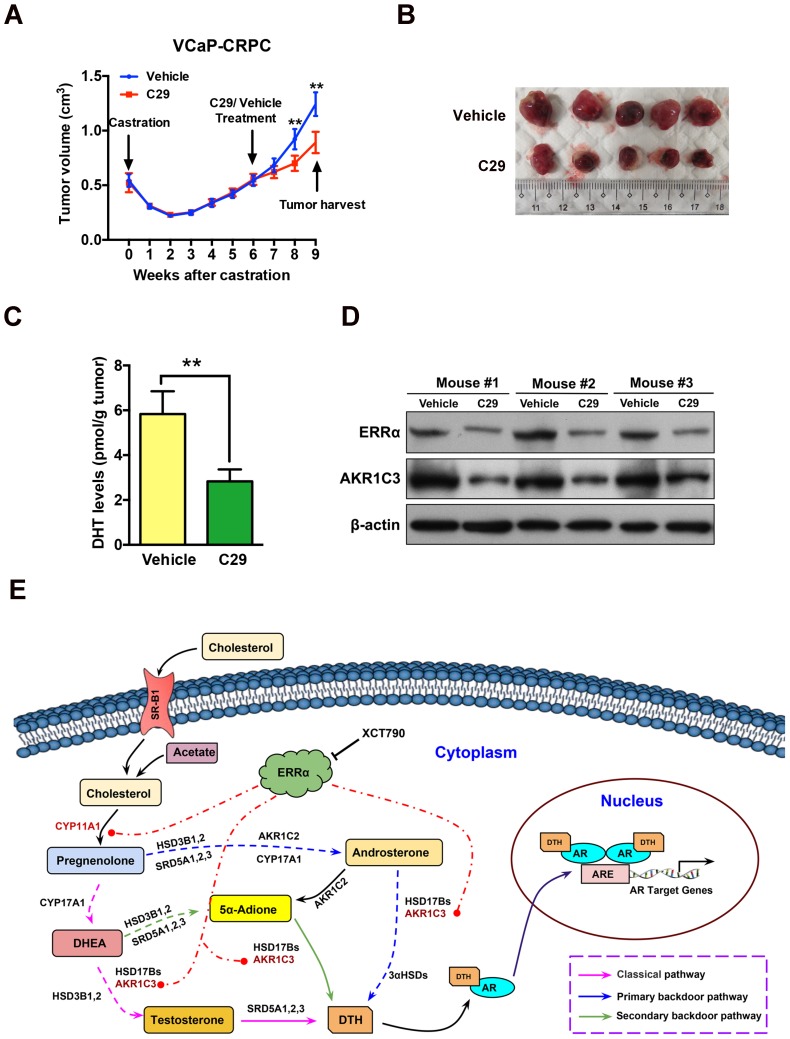
Enhanced intratumoral androgen biosynthesis and persistent androgen receptor (AR) signaling are key factors responsible for the relapse growth of castration-resistant prostate cancer (CRPC). Residual intraprostatic androgens can be produced by de novo synthesis of androgens from cholesterol or conversion from adrenal androgens by steroidogenic enzymes expressed in prostate cancer cells via different steroidogenic pathways. However, the dysregulation of androgen biosynthetic enzymes in CRPC still remains poorly understood. This study aims to elucidate the role of the nuclear receptor, estrogen-related receptor alpha (ERRα, ESRRA), in the promotion of androgen biosynthesis in CRPC growth. Methods: ERRα expression in CRPC patients was analyzed using Gene Expression Omnibus (GEO) datasets and validated in established CRPC xenograft model. The roles of ERRα in the promotion of castration-resistant growth were elucidated by overexpression and knockdown studies and the intratumoral androgen levels were measured by UPLC-MS/MS. The effect of suppression of ERRα activity in the potentiation of sensitivity to androgen-deprivation was determined using an ERRα inverse agonist. Results: ERRα exhibited an increased expression in metastatic CRPC and CRPC xenograft model, could act to promote castration-resistant growth via direct transactivation of two key androgen synthesis enzymes CYP11A1 and AKR1C3, and hence enhance intraprostatic production of dihydrotestosterone (DHT) and activation of AR signaling in prostate cancer cells. Notably, inhibition of ERRα activity by an inverse agonist XCT790 could reduce the DHT production and suppress AR signaling in prostate cancer cells. Conclusion: Our study reveals a new role of ERRα in the intratumoral androgen biosynthesis in CRPC via its transcriptional control of steroidogenic enzymes, and also provides a novel insight that targeting ERRα could be a potential androgen-deprivation strategy for the management of CRPC.
Keywords: AKR1C3; CYP11A1; ERRα; castration resistance; intratumoral steroidogenesis; nuclear receptor; prostate cancer.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Nadiminty N, Gao AC. Mechanisms of persistent activation of the androgen receptor in CRPC: recent advances and future perspectives. World J Urol. 2012;30:287–95. - PubMed
-
- Egan A, Dong Y, Zhang H, Qi Y, Balk SP, Sartor O. Castration-resistant prostate cancer: adaptive responses in the androgen axis. Cancer Treat Rev. 2014;40:426–33. - PubMed
-
- Mitsiades N. A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res. 2013;73:4599–605. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
