

Hypertrophic Cardiomyopathy With Left Ventricular Systolic Dysfunction: Insights From the SHaRe Registry
- PMID: 32228044
- PMCID: PMC7182243
- DOI: 10.1161/CIRCULATIONAHA.119.044366
Hypertrophic Cardiomyopathy With Left Ventricular Systolic Dysfunction: Insights From the SHaRe Registry
Abstract
Background: The term "end stage" has been used to describe hypertrophic cardiomyopathy (HCM) with left ventricular systolic dysfunction (LVSD), defined as occurring when left ventricular ejection fraction is <50%. The prognosis of HCM-LVSD has reportedly been poor, but because of its relative rarity, the natural history remains incompletely characterized.
Methods: Data from 11 high-volume HCM specialty centers making up the international SHaRe Registry (Sarcomeric Human Cardiomyopathy Registry) were used to describe the natural history of patients with HCM-LVSD. Cox proportional hazards models were used to identify predictors of prognosis and incident development.
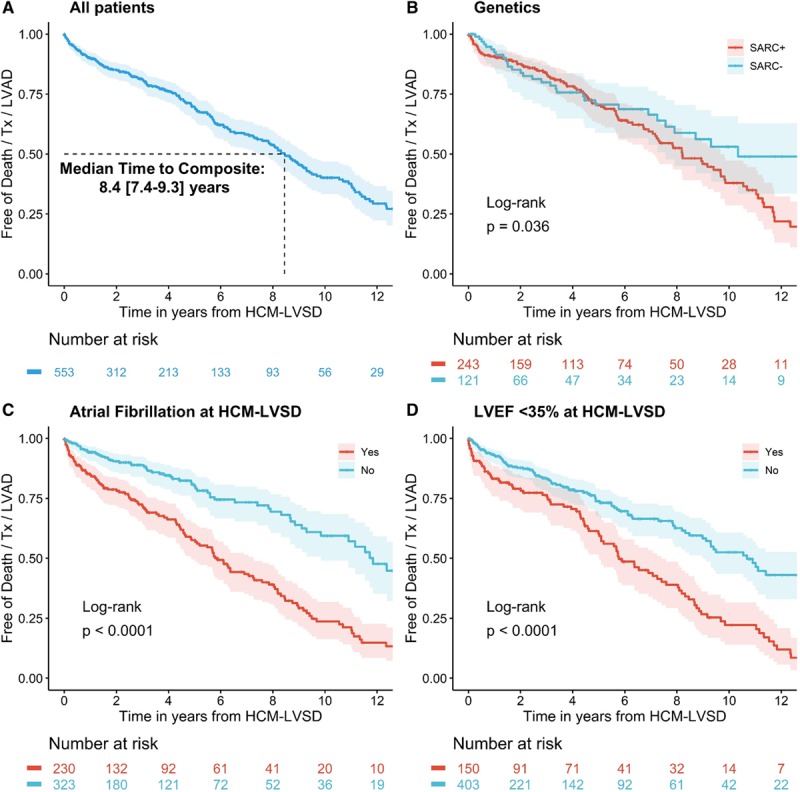
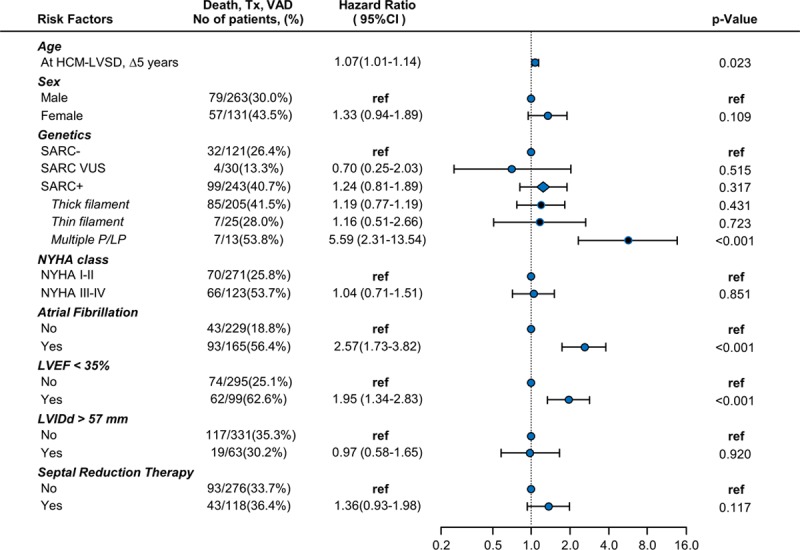
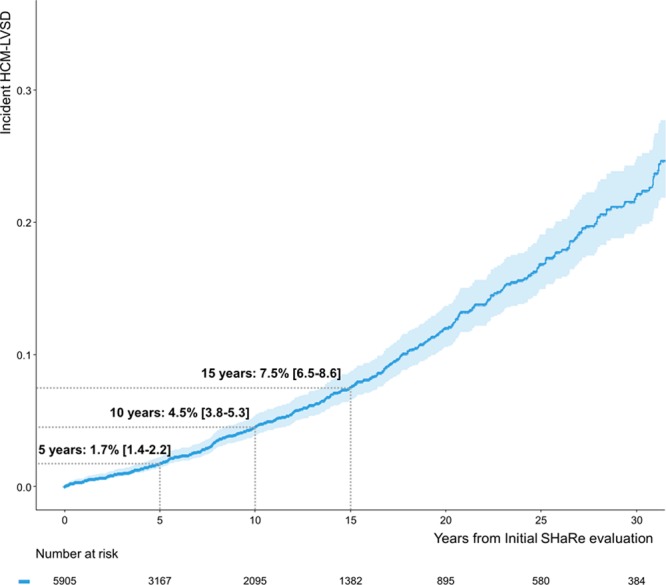
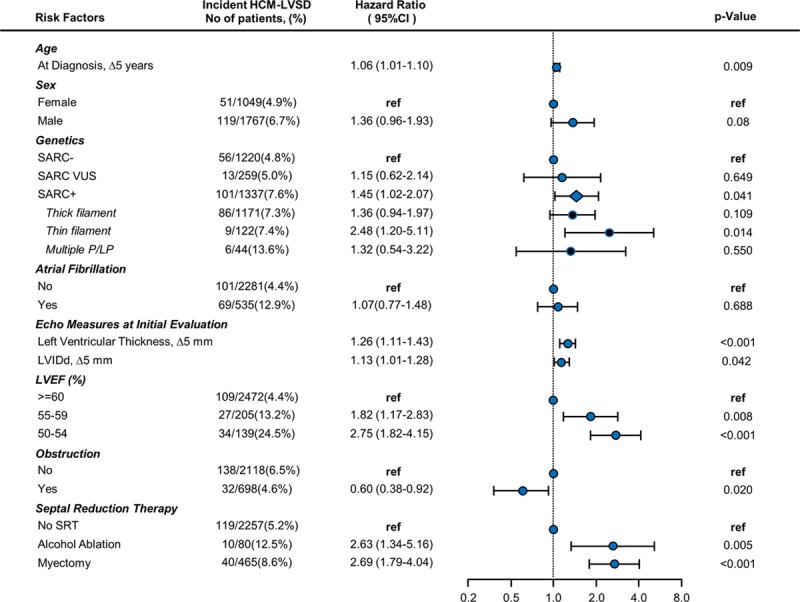
Results: From a cohort of 6793 patients with HCM, 553 (8%) met the criteria for HCM-LVSD. Overall, 75% of patients with HCM-LVSD experienced clinically relevant events, and 35% met the composite outcome (all-cause death [n=128], cardiac transplantation [n=55], or left ventricular assist device implantation [n=9]). After recognition of HCM-LVSD, the median time to composite outcome was 8.4 years. However, there was substantial individual variation in natural history. Significant predictors of the composite outcome included the presence of multiple pathogenic/likely pathogenic sarcomeric variants (hazard ratio [HR], 5.6 [95% CI, 2.3-13.5]), atrial fibrillation (HR, 2.6 [95% CI, 1.7-3.5]), and left ventricular ejection fraction <35% (HR, 2.0 [95% CI, 1.3-2.8]). The incidence of new HCM-LVSD was ≈7.5% over 15 years. Significant predictors of developing incident HCM-LVSD included greater left ventricular cavity size (HR, 1.1 [95% CI, 1.0-1.3] and wall thickness (HR, 1.3 [95% CI, 1.1-1.4]), left ventricular ejection fraction of 50% to 60% (HR, 1.8 [95% CI, 1.2, 2.8]-2.8 [95% CI, 1.8-4.2]) at baseline evaluation, the presence of late gadolinium enhancement on cardiac magnetic resonance imaging (HR, 2.3 [95% CI, 1.0-4.9]), and the presence of a pathogenic/likely pathogenic sarcomeric variant, particularly in thin filament genes (HR, 1.5 [95% CI, 1.0-2.1] and 2.5 [95% CI, 1.2-5.1], respectively).
Conclusions: HCM-LVSD affects ≈8% of patients with HCM. Although the natural history of HCM-LVSD was variable, 75% of patients experienced adverse events, including 35% experiencing a death equivalent an estimated median time of 8.4 years after developing systolic dysfunction. In addition to clinical features, genetic substrate appears to play a role in both prognosis (multiple sarcomeric variants) and the risk for incident development of HCM-LVSD (thin filament variants).
Keywords: cardiomyopathy, hypertrophic; genetics; heart failure; prognosis; ventricular dysfunction.
Figures
References
-
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–2779. - PubMed
-
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796. doi: 10.1161/CIR.0b013e318223e230.
-
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. doi: 10.1016/j.jacc.2015.01.019. - PubMed
-
- Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–888. doi: 10.1038/gim.2014.205. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
