

Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival
- PMID: 32228719
- PMCID: PMC7106909
- DOI: 10.1186/s13073-020-00729-2
Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival
Abstract
Background: Tumor mutational burden (TMB; the quantity of aberrant nucleotide sequences a given tumor may harbor) has been associated with response to immune checkpoint inhibitor therapy and is gaining broad acceptance as a result. However, TMB harbors intrinsic variability across cancer types, and its assessment and interpretation are poorly standardized.
Methods: Using a standardized approach, we quantify the robustness of TMB as a metric and its potential as a predictor of immunotherapy response and survival among a diverse cohort of cancer patients. We also explore the additive predictive potential of RNA-derived variants and neoepitope burden, incorporating several novel metrics of immunogenic potential.
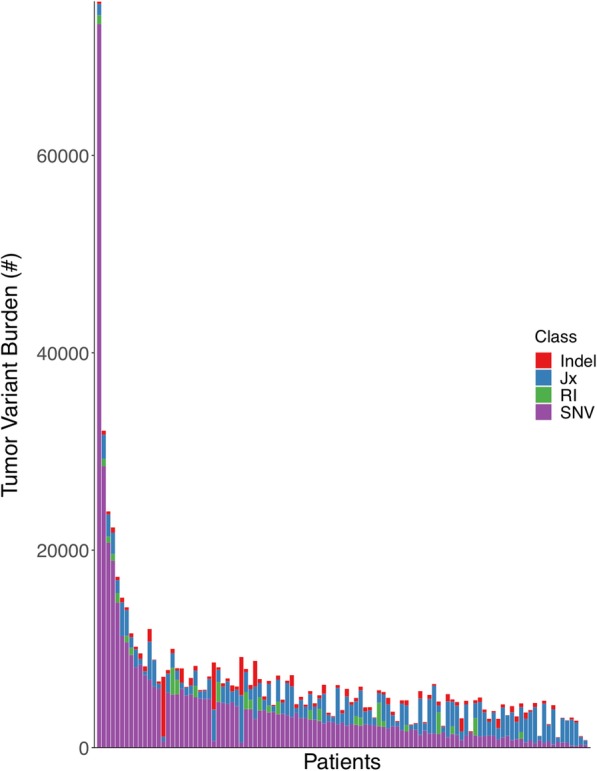
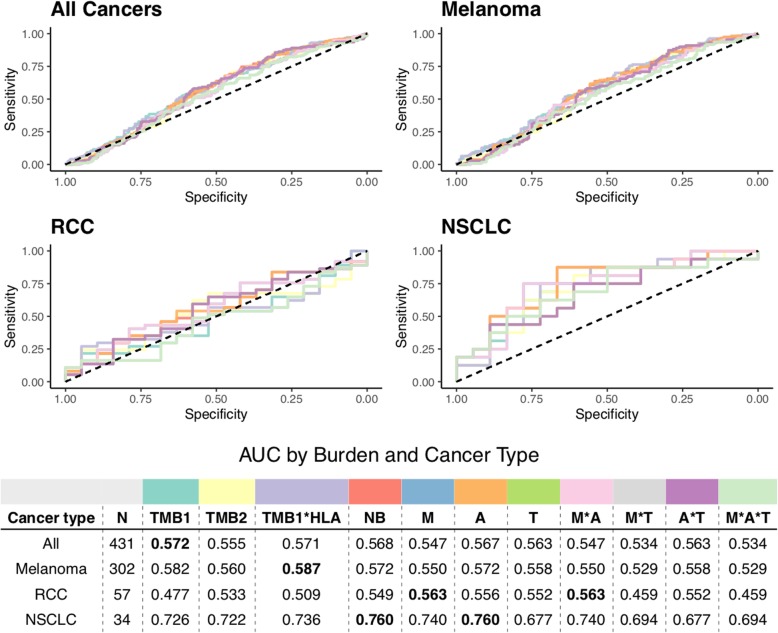
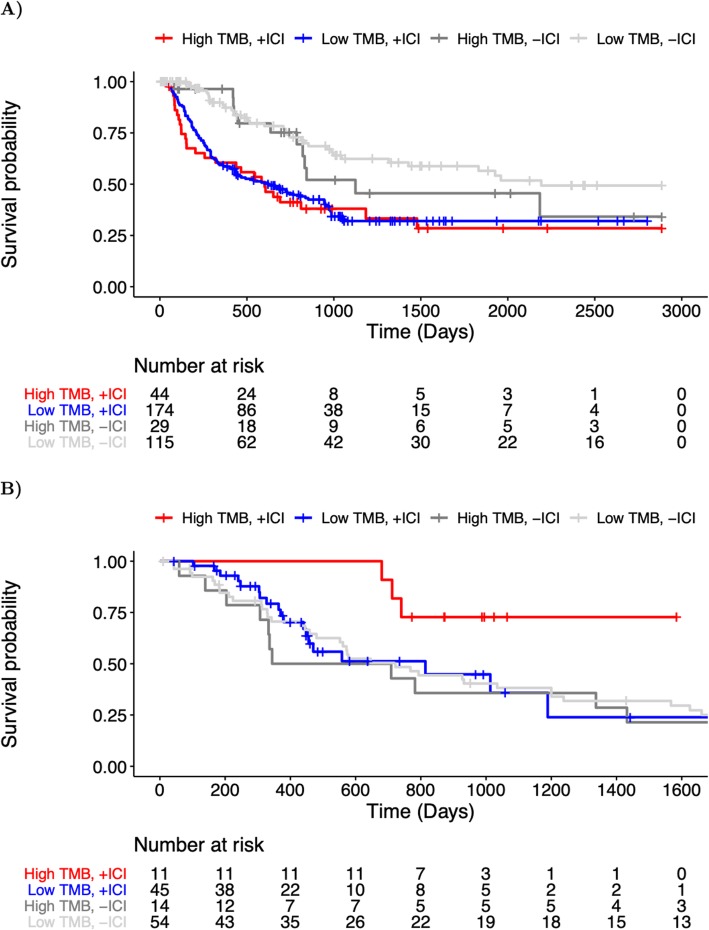
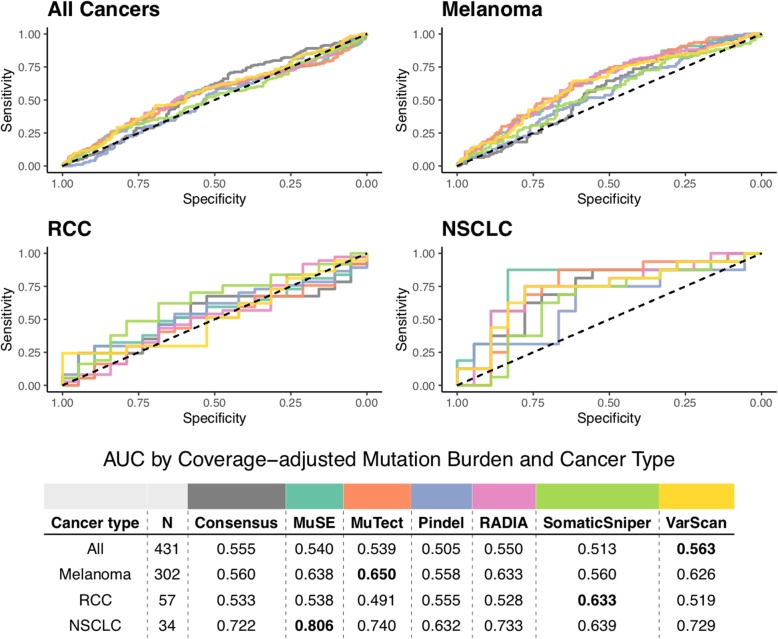
Results: We find that TMB is a partial predictor of immunotherapy response in melanoma and non-small cell lung cancer, but not renal cell carcinoma. We find that TMB is predictive of overall survival in melanoma patients receiving immunotherapy, but not in an immunotherapy-naive population. We also find that it is an unstable metric with potentially problematic repercussions for clinical cohort classification. We finally note minimal additional predictive benefit to assessing neoepitope burden or its bulk derivatives, including RNA-derived sources of neoepitopes.
Conclusions: We find sufficient cause to suggest that the predictive clinical value of TMB should not be overstated or oversimplified. While it is readily quantified, TMB is at best a limited surrogate biomarker of immunotherapy response. The data do not support isolated use of TMB in renal cell carcinoma.
Keywords: Immunotherapy response; Neoantigens; Neoepitope burden; Neoepitopes; Retained introns; Splice junctions; TMB; Tumor mutational burden; Tumor variant burden.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
