

Defining metrics for whole-genome sequence analysis of MRSA in clinical practice
- PMID: 32228804
- PMCID: PMC7276698
- DOI: 10.1099/mgen.0.000354
Defining metrics for whole-genome sequence analysis of MRSA in clinical practice
Abstract
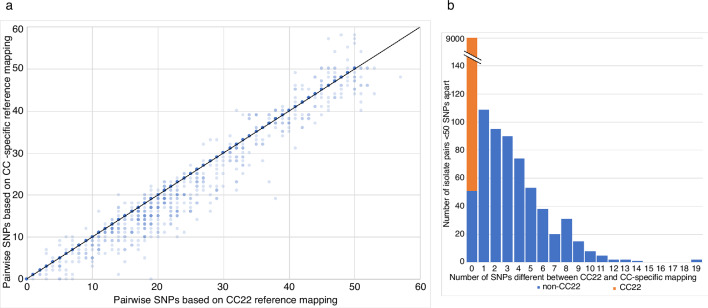
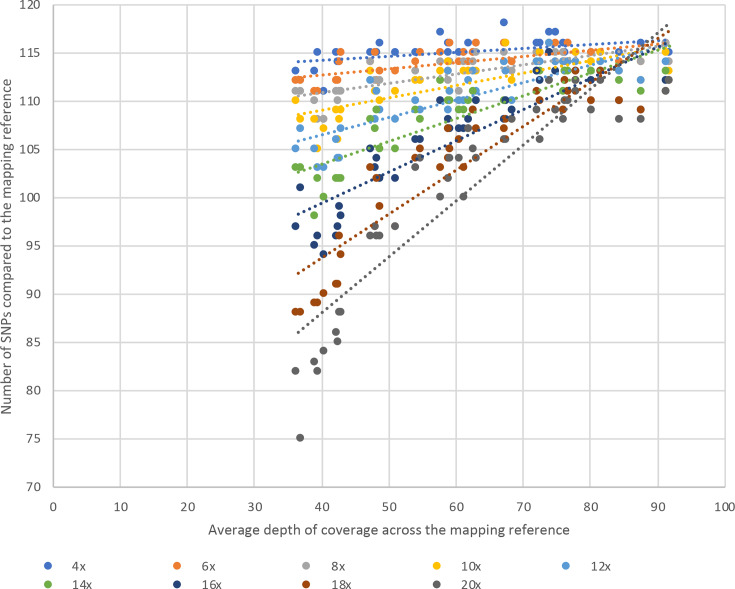
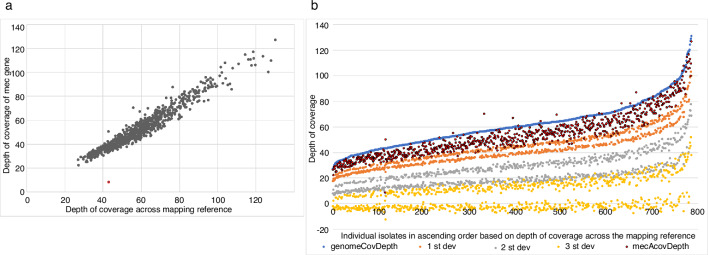
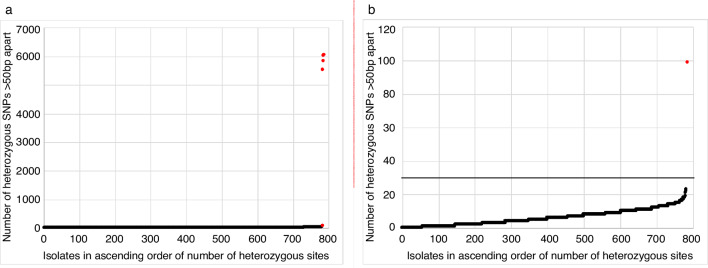
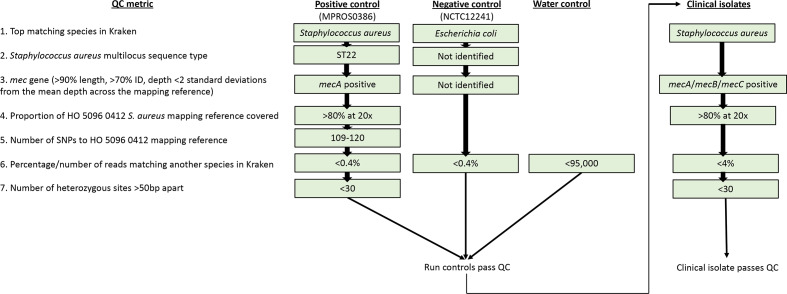
Bacterial sequencing will become increasingly adopted in routine microbiology laboratories. Here, we report the findings of a technical evaluation of almost 800 clinical methicillin-resistant Staphylococcus aureus (MRSA) isolates, in which we sought to define key quality metrics to support MRSA sequencing in clinical practice. We evaluated the accuracy of mapping to a generic reference versus clonal complex (CC)-specific mapping, which is more computationally challenging. Focusing on isolates that were genetically related (<50 single nucleotide polymorphisms (SNPs)) and belonged to prevalent sequence types, concordance between these methods was 99.5 %. We use MRSA MPROS0386 to control for base calling accuracy by the sequencer, and used multiple repeat sequences of the control to define a permitted range of SNPs different to the mapping reference for this control (equating to 3 standard deviations from the mean). Repeat sequences of the control were also used to demonstrate that SNP calling was most accurate across differing coverage depths (above 35×, the lowest depth in our study) when the depth required to call a SNP as present was at least 4-8×. Using 786 MRSA sequences, we defined a robust measure for mec gene detection to reduce false-positives arising from contamination, which was no greater than 2 standard deviations below the average depth of coverage across the genome. Sequencing from bacteria harvested from clinical plates runs an increased risk of contamination with the same or different species, and we defined a cut-off of 30 heterozygous sites >50 bp apart to identify same-species contamination for MRSA. These metrics were combined into a quality-control (QC) flowchart to determine whether sequence runs and individual clinical isolates passed QC, which could be adapted by future automated analysis systems to enable rapid hands-off sequence analysis by clinical laboratories.
Keywords: MRSA; quality metrics; translational; whole-genome sequencing.
Conflict of interest statement
S.J.P., J.P. and F.C. are consultants to Next Gen Diagnostics, and S.J.P. is a consultant to Specific Technologies. E.B. is an employee of Next Gen Diagnostics. The other authors have no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous