A Sample-to-Report Solution for Taxonomic Identification of Cultured Bacteria in the Clinical Setting Based on Nanopore Sequencing
- PMID: 32229603
- PMCID: PMC7269405
- DOI: 10.1128/JCM.00060-20
A Sample-to-Report Solution for Taxonomic Identification of Cultured Bacteria in the Clinical Setting Based on Nanopore Sequencing
Abstract
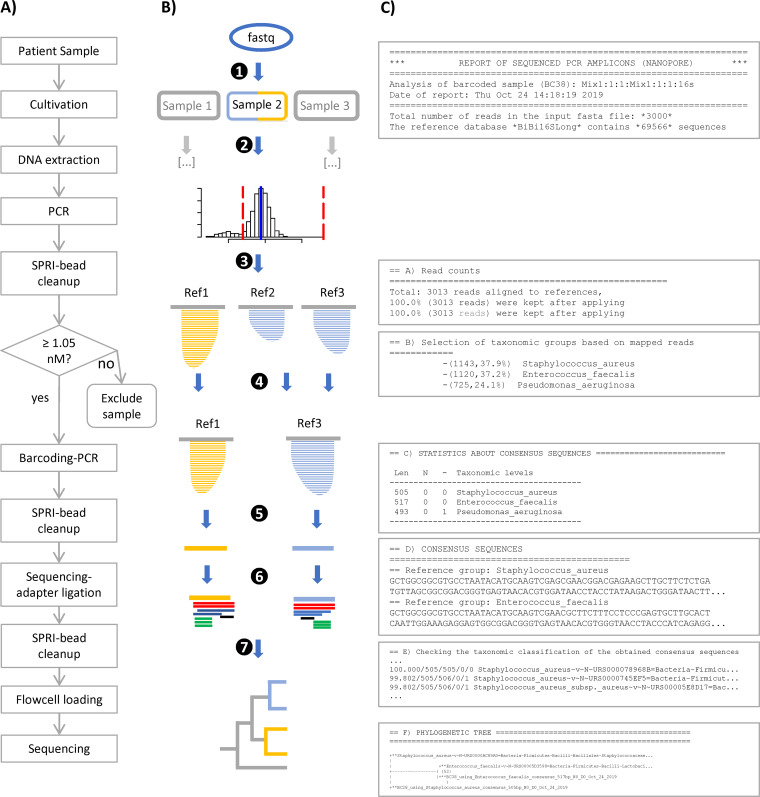
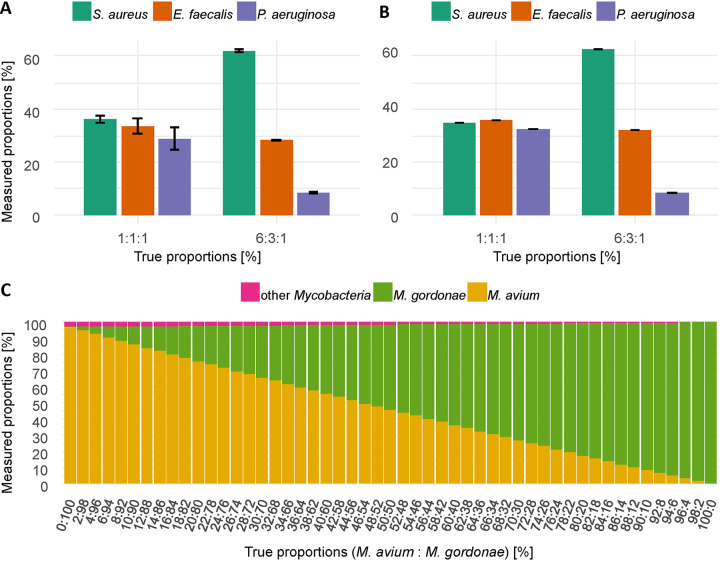
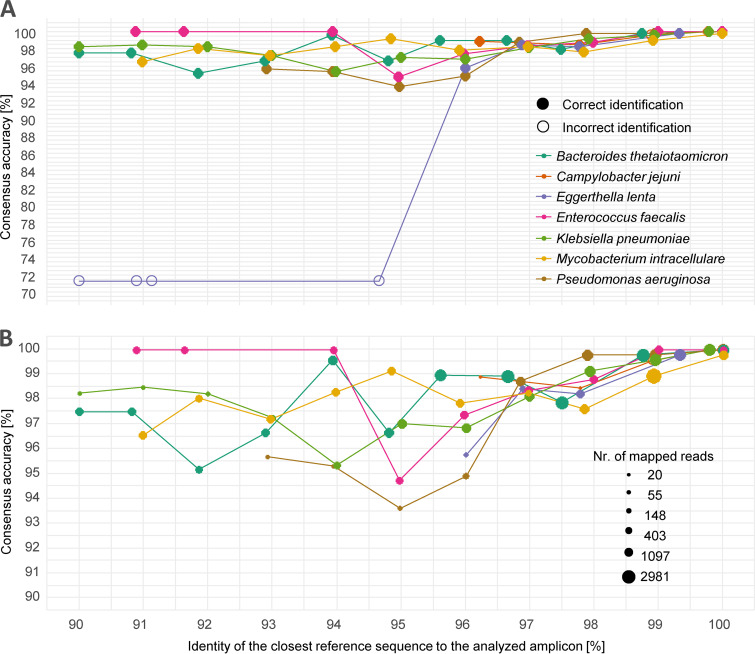
Amplicon sequencing of the 16S rRNA gene is commonly used for the identification of bacterial isolates in diagnostic laboratories and mostly relies on the Sanger sequencing method. The latter, however, suffers from a number of limitations, with the most significant being the inability to resolve mixed amplicons when closely related species are coamplified from a mixed culture. This often leads to either increased turnaround time or absence of usable sequence data. Short-read next-generation sequencing (NGS) technologies could solve the mixed amplicon issue but would lack both cost efficiency at low throughput and fast turnaround times. Nanopore sequencing developed by Oxford Nanopore Technologies (ONT) could solve those issues by enabling a flexible number of samples per run and an adjustable sequencing time. Here, we report on the development of a standardized laboratory workflow combined with a fully automated analysis pipeline LORCAN (long read consensus analysis), which together provide a sample-to-report solution for amplicon sequencing and taxonomic identification of the resulting consensus sequences. Validation of the approach was conducted on a panel of reference strains and on clinical samples consisting of single or mixed rRNA amplicons associated with various bacterial genera by direct comparison to the corresponding Sanger sequences. Additionally, simulated read and amplicon mixtures were used to assess LORCAN's behavior when dealing with samples with known cross-contamination levels. We demonstrate that by combining ONT amplicon sequencing results with LORCAN, the accuracy of Sanger sequencing can be closely matched (>99.6% sequence identity) and that mixed samples can be resolved at the single-base resolution level. The presented approach has the potential to significantly improve the flexibility, reliability, and availability of amplicon sequencing in diagnostic settings.
Keywords: 16S RNA gene; bioinformatics; clinical methods; diagnostics; nanopore; sequencing; taxonomy.
Copyright © 2020 Neuenschwander et al.
Figures