

Single-cell RNA sequencing in cardiovascular development, disease and medicine
- PMID: 32231331
- PMCID: PMC7528042
- DOI: 10.1038/s41569-020-0359-y
Single-cell RNA sequencing in cardiovascular development, disease and medicine
Abstract
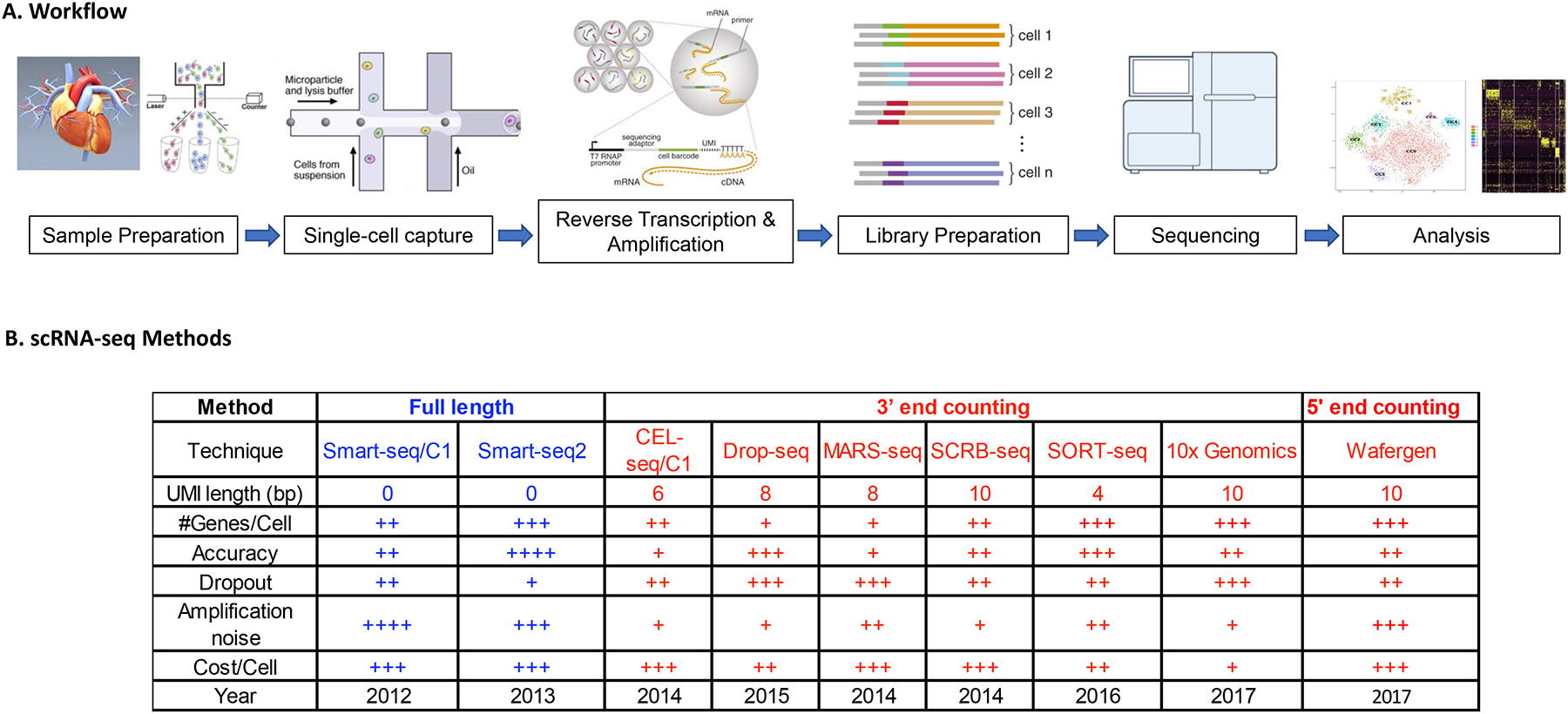
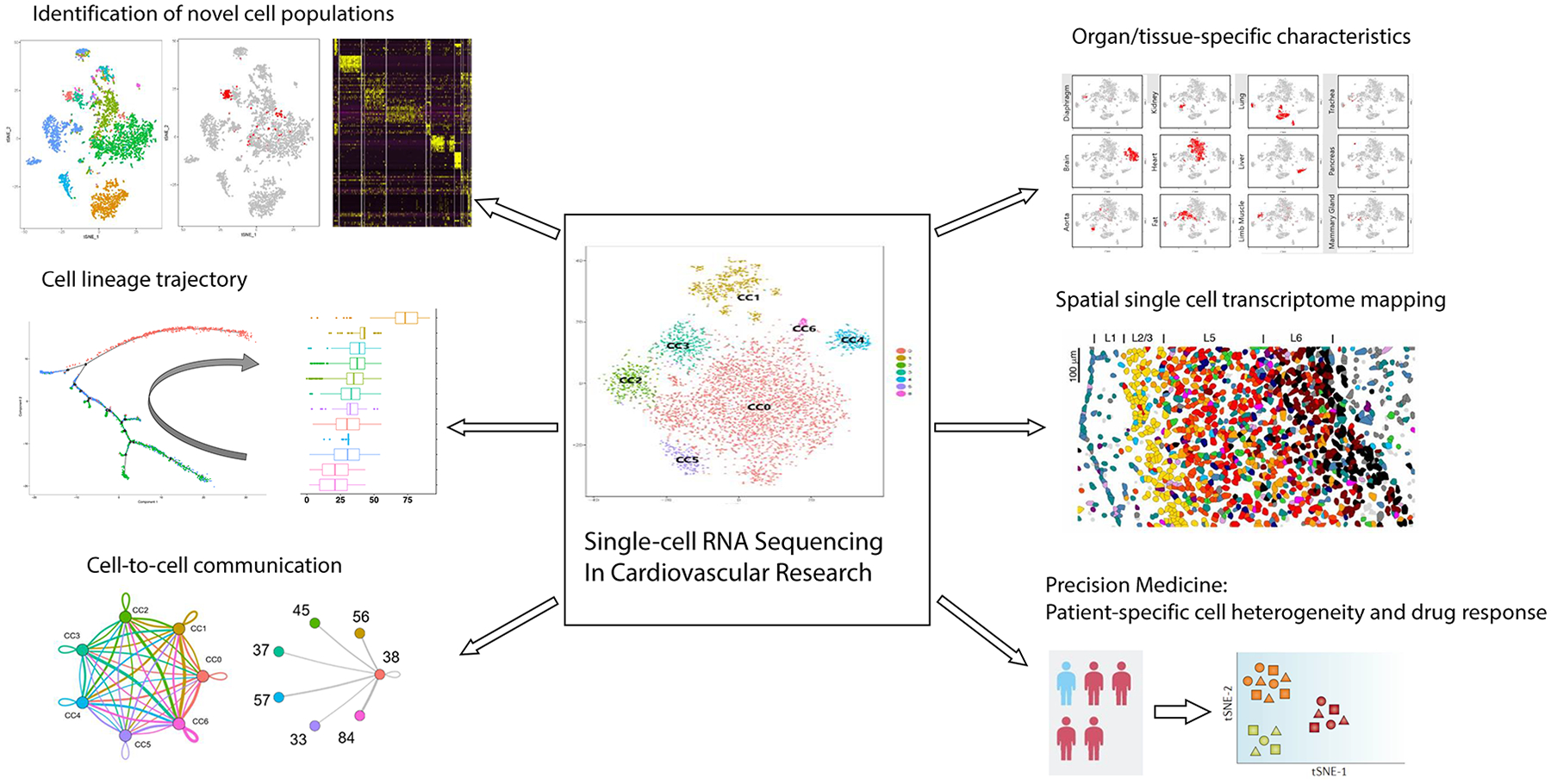
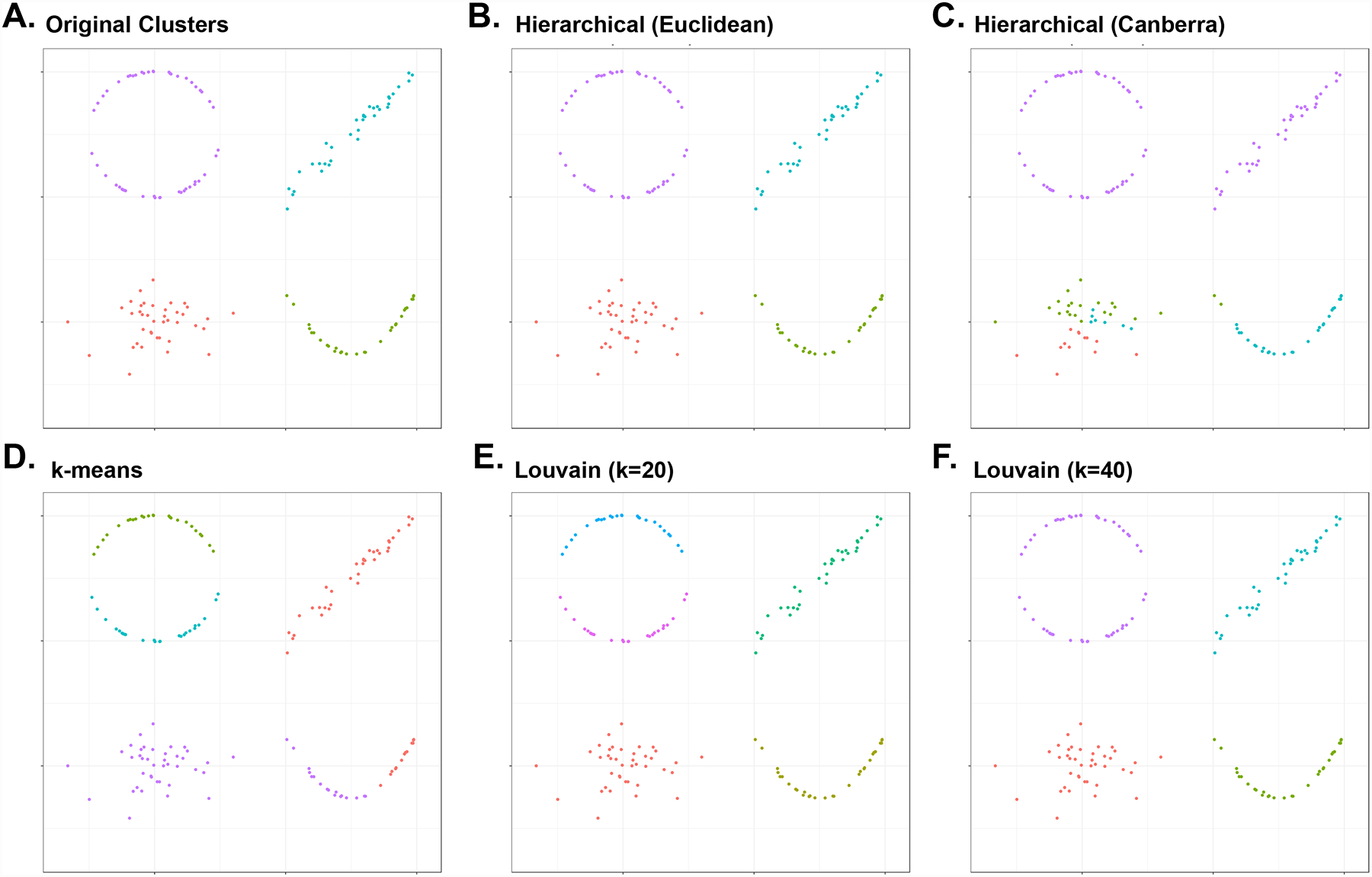
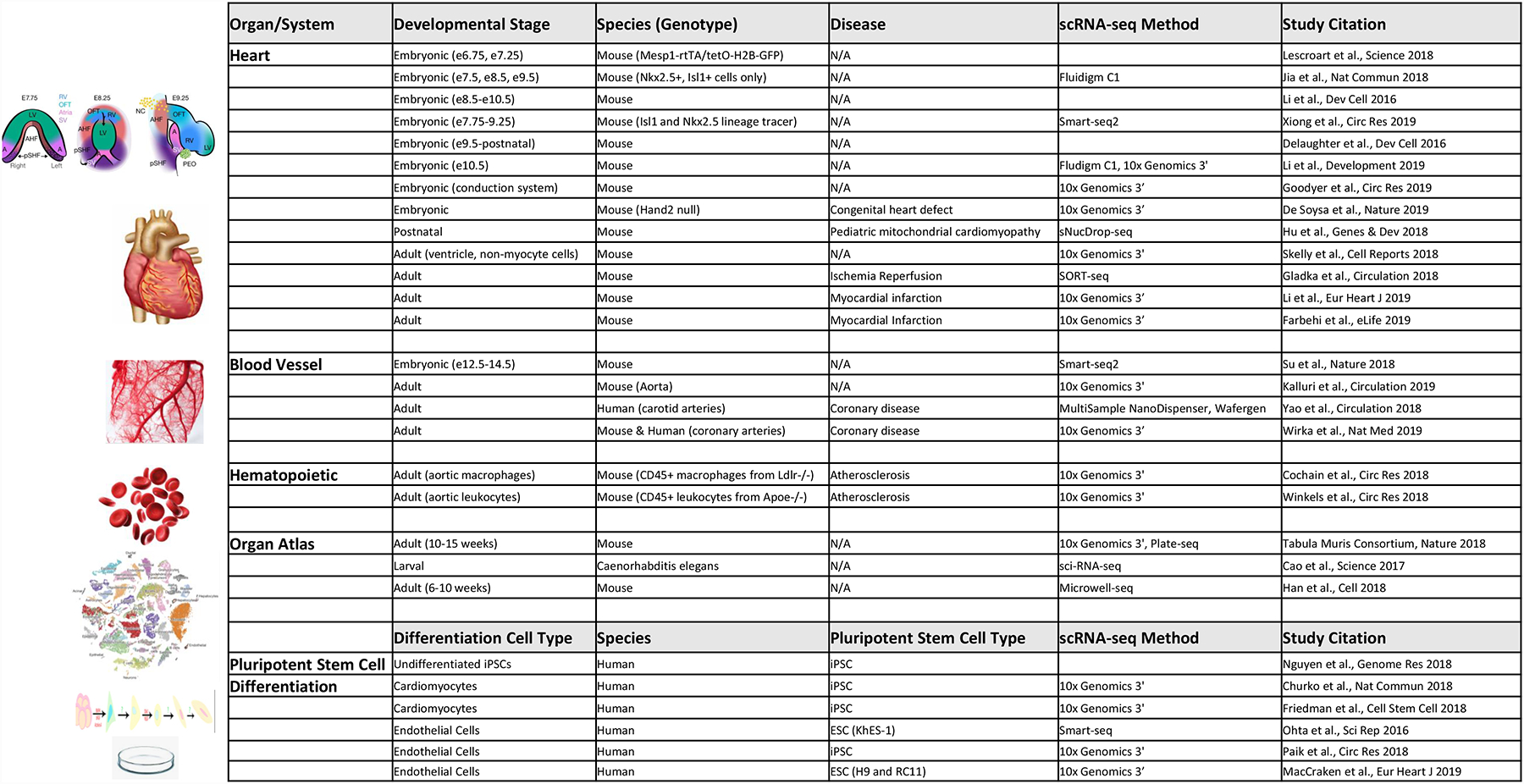
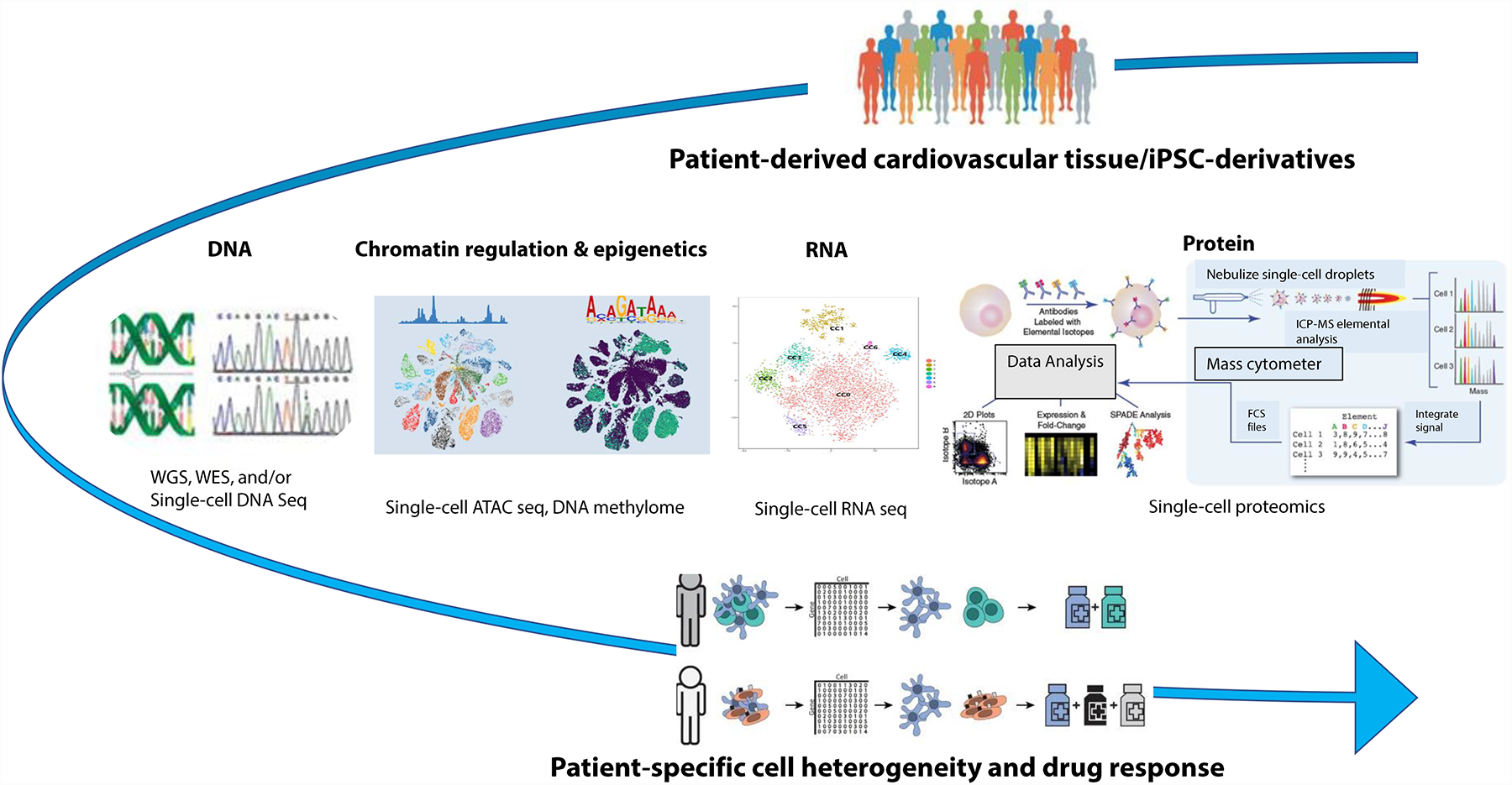
Advances in single-cell RNA sequencing (scRNA-seq) technologies in the past 10 years have had a transformative effect on biomedical research, enabling the profiling and analysis of the transcriptomes of single cells at unprecedented resolution and throughput. Specifically, scRNA-seq has facilitated the identification of novel or rare cell types, the analysis of single-cell trajectory construction and stem or progenitor cell differentiation, and the comparison of healthy and disease-related tissues at single-cell resolution. These applications have been critical in advances in cardiovascular research in the past decade as evidenced by the generation of cell atlases of mammalian heart and blood vessels and the elucidation of mechanisms involved in cardiovascular development and stem or progenitor cell differentiation. In this Review, we summarize the currently available scRNA-seq technologies and analytical tools and discuss the latest findings using scRNA-seq that have substantially improved our knowledge on the development of the cardiovascular system and the mechanisms underlying cardiovascular diseases. Furthermore, we examine emerging strategies that integrate multimodal single-cell platforms, focusing on future applications in cardiovascular precision medicine that use single-cell omics approaches to characterize cell-specific responses to drugs or environmental stimuli and to develop effective patient-specific therapeutics.
Conflict of interest statement
Competing interests
H.Y.C. is a co-founder of Accent Therapeutics and Boundless Bio, and an advisor to 10× Genomics, Arsenal Biosciences and Spring Discovery. J.C.W. is a co-founder of Khloris Biosciences, but has no competing interests, as the work presented here is completely independent. The other authors declare no competing interests.
Figures
References
-
- Heid CA, Stevens J, Livak KJ & Williams PM Real time quantitative PCR. Genome Res. 6, 986–994 (1996). - PubMed
-
- Islam S et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 11, 163–166 (2014). - PubMed
-
- Shapiro E, Biezuner T & Linnarsson S Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet 14, 618–630 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
