

Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing
- PMID: 32231336
- PMCID: PMC7416462
- DOI: 10.1038/s41587-020-0470-y
Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing
Abstract
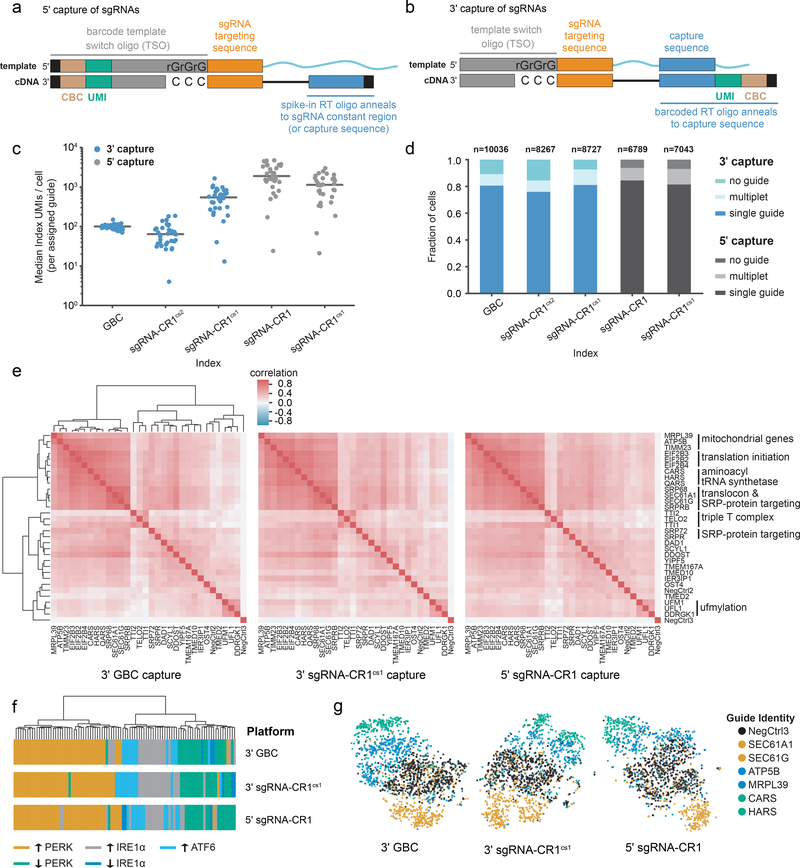
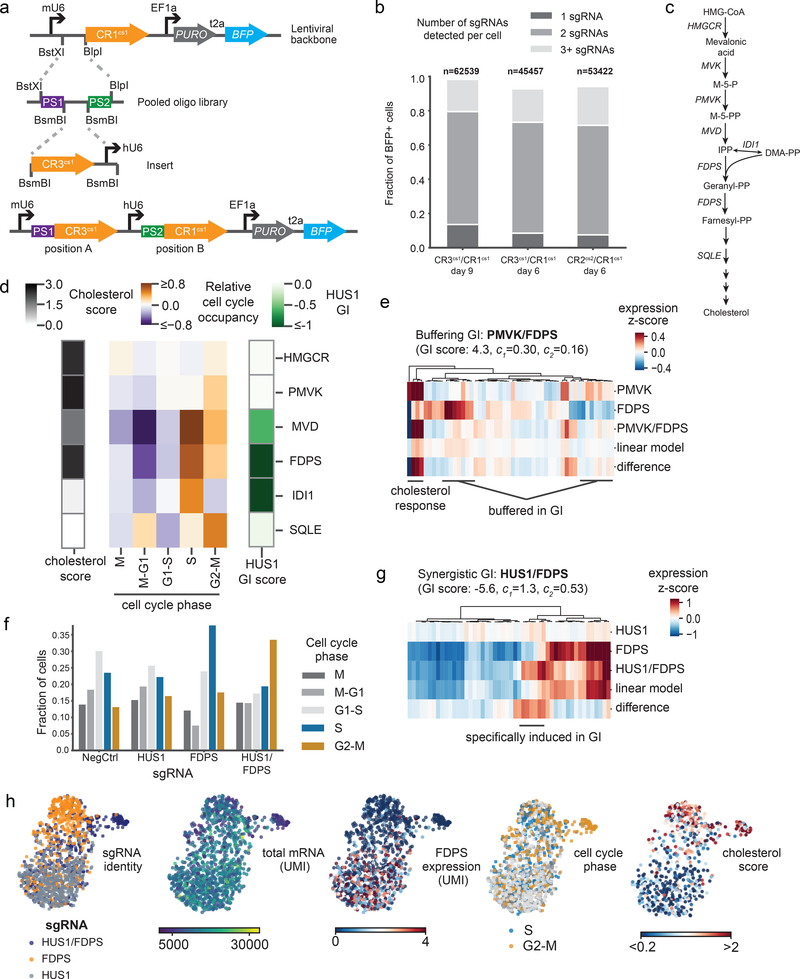
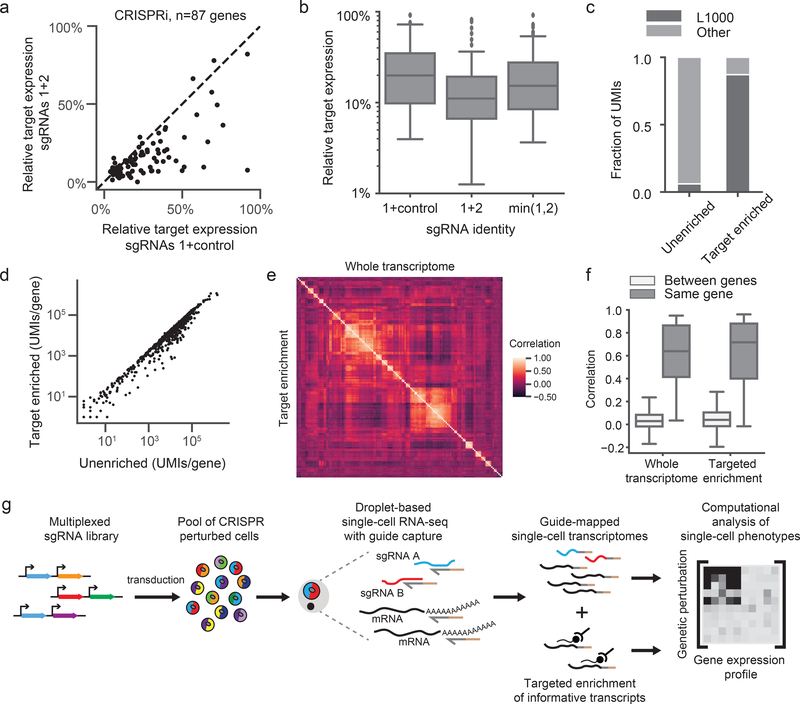
Single-cell CRISPR screens enable the exploration of mammalian gene function and genetic regulatory networks. However, use of this technology has been limited by reliance on indirect indexing of single-guide RNAs (sgRNAs). Here we present direct-capture Perturb-seq, a versatile screening approach in which expressed sgRNAs are sequenced alongside single-cell transcriptomes. Direct-capture Perturb-seq enables detection of multiple distinct sgRNA sequences from individual cells and thus allows pooled single-cell CRISPR screens to be easily paired with combinatorial perturbation libraries that contain dual-guide expression vectors. We demonstrate the utility of this approach for high-throughput investigations of genetic interactions and, leveraging this ability, dissect epistatic interactions between cholesterol biogenesis and DNA repair. Using direct capture Perturb-seq, we also show that targeting individual genes with multiple sgRNAs per cell improves efficacy of CRISPR interference and activation, facilitating the use of compact, highly active CRISPR libraries for single-cell screens. Last, we show that hybridization-based target enrichment permits sensitive, specific sequencing of informative transcripts from single-cell RNA-seq experiments.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
