

Pulmonary Pathogens Adapt to Immune Signaling Metabolites in the Airway
- PMID: 32231665
- PMCID: PMC7082326
- DOI: 10.3389/fimmu.2020.00385
Pulmonary Pathogens Adapt to Immune Signaling Metabolites in the Airway
Abstract
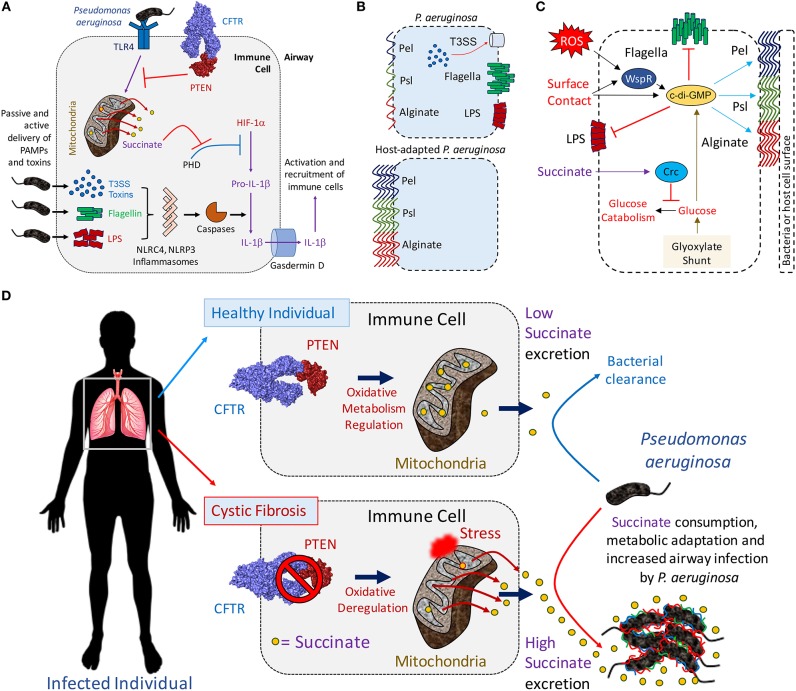
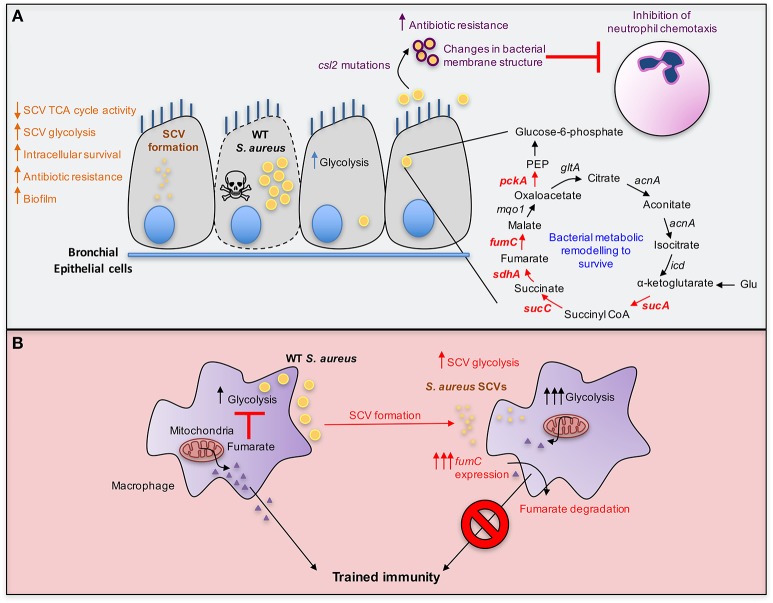
A limited number of pulmonary pathogens are able to evade normal mucosal defenses to establish acute infection and then adapt to cause chronic pneumonias. Pathogens, such as Pseudomonas aeruginosa or Staphylococcus aureus, are typically associated with infection in patients with underlying pulmonary disease or damage, such as cystic fibrosis (CF) or chronic obstructive pulmonary disease (COPD). To establish infection, bacteria express a well-defined set of so-called virulence factors that facilitate colonization and activate an immune response, gene products that have been identified in murine models. Less well-understood are the adaptive changes that occur over time in vivo, enabling the organisms to evade innate and adaptive immune clearance mechanisms. These colonizers proliferate, generating a population sufficient to provide selection for mutants, such as small colony variants and mucoid variants, that are optimized for long term infection. Such host-adapted strains have evolved in response to selective pressure such as antibiotics and the recruitment of phagocytes at sites of infection and their release of signaling metabolites (e.g., succinate). These metabolites can potentially function as substrates for bacterial growth and but also generate oxidant stress. Whole genome sequencing and quantified expression of selected genes have helped to explain how P. aeruginosa and S. aureus adapt to the presence of these metabolites over the course of in vivo infection. The serial isolation of clonally related strains from patients with cystic fibrosis has provided the opportunity to identify bacterial metabolic pathways that are altered under this immune pressure, such as the anti-oxidant glyoxylate and pentose phosphate pathways, routes contributing to the generation of biofilms. These metabolic pathways and biofilm itself enable the organisms to dissipate oxidant stress, while providing protection from phagocytosis. Stimulation of host immune signaling metabolites by these pathogens drives bacterial adaptation and promotes their persistence in the airways. The inherent metabolic flexibility of P. aeruginosa and S. aureus is a major factor in their success as pulmonary pathogens.
Keywords: COPD; Pseudomonas aeruginosa; Staphylococcus aereus; cystic fibrosis; fumarate; immunometabolism; inflammation; succinate.
Copyright © 2020 Riquelme, Wong Fok Lung and Prince.
Figures
Similar articles
-
Immunometabolites Drive Bacterial Adaptation to the Airway.Front Immunol. 2021 Nov 25;12:790574. doi: 10.3389/fimmu.2021.790574. eCollection 2021. Front Immunol. 2021. PMID: 34899759 Free PMC article. Review.
-
Association of Diverse Staphylococcus aureus Populations with Pseudomonas aeruginosa Coinfection and Inflammation in Cystic Fibrosis Airway Infection.mSphere. 2021 Jun 30;6(3):e0035821. doi: 10.1128/mSphere.00358-21. Epub 2021 Jun 23. mSphere. 2021. PMID: 34160233 Free PMC article.
-
The Small RNA ErsA Plays a Role in the Regulatory Network of Pseudomonas aeruginosa Pathogenicity in Airway Infections.mSphere. 2020 Oct 14;5(5):e00909-20. doi: 10.1128/mSphere.00909-20. mSphere. 2020. PMID: 33055260 Free PMC article.
-
Adaptation of Pseudomonas aeruginosa in Cystic Fibrosis airways influences virulence of Staphylococcus aureus in vitro and murine models of co-infection.PLoS One. 2014 Mar 6;9(3):e89614. doi: 10.1371/journal.pone.0089614. eCollection 2014. PLoS One. 2014. PMID: 24603807 Free PMC article.
-
Airway immunometabolites fuel Pseudomonas aeruginosa infection.Respir Res. 2020 Dec 10;21(1):326. doi: 10.1186/s12931-020-01591-x. Respir Res. 2020. PMID: 33302964 Free PMC article. Review.
Cited by
-
How Bacterial Adaptation to Cystic Fibrosis Environment Shapes Interactions Between Pseudomonas aeruginosa and Staphylococcus aureus.Front Microbiol. 2021 Mar 3;12:617784. doi: 10.3389/fmicb.2021.617784. eCollection 2021. Front Microbiol. 2021. PMID: 33746915 Free PMC article. Review.
-
Staphylococcus aureus adaptive evolution: Recent insights on how immune evasion, immunometabolic subversion and host genetics impact vaccine development.Front Cell Infect Microbiol. 2022 Dec 27;12:1060810. doi: 10.3389/fcimb.2022.1060810. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 36636720 Free PMC article. Review.
-
Trained immunity: A "new" weapon in the fight against infectious diseases.Front Immunol. 2023 Mar 13;14:1147476. doi: 10.3389/fimmu.2023.1147476. eCollection 2023. Front Immunol. 2023. PMID: 36993966 Free PMC article. Review.
-
Metabolic Modeling to Interrogate Microbial Disease: A Tale for Experimentalists.Front Mol Biosci. 2021 Feb 18;8:634479. doi: 10.3389/fmolb.2021.634479. eCollection 2021. Front Mol Biosci. 2021. PMID: 33681294 Free PMC article. Review.
-
PmiR senses 2-methylisocitrate levels to regulate bacterial virulence in Pseudomonas aeruginosa.Sci Adv. 2022 Dec 9;8(49):eadd4220. doi: 10.1126/sciadv.add4220. Epub 2022 Dec 7. Sci Adv. 2022. PMID: 36475801 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources