

The hidden enzymology of bacterial natural product biosynthesis
- PMID: 32232178
- PMCID: PMC7104373
- DOI: 10.1038/s41570-019-0107-1
The hidden enzymology of bacterial natural product biosynthesis
Abstract
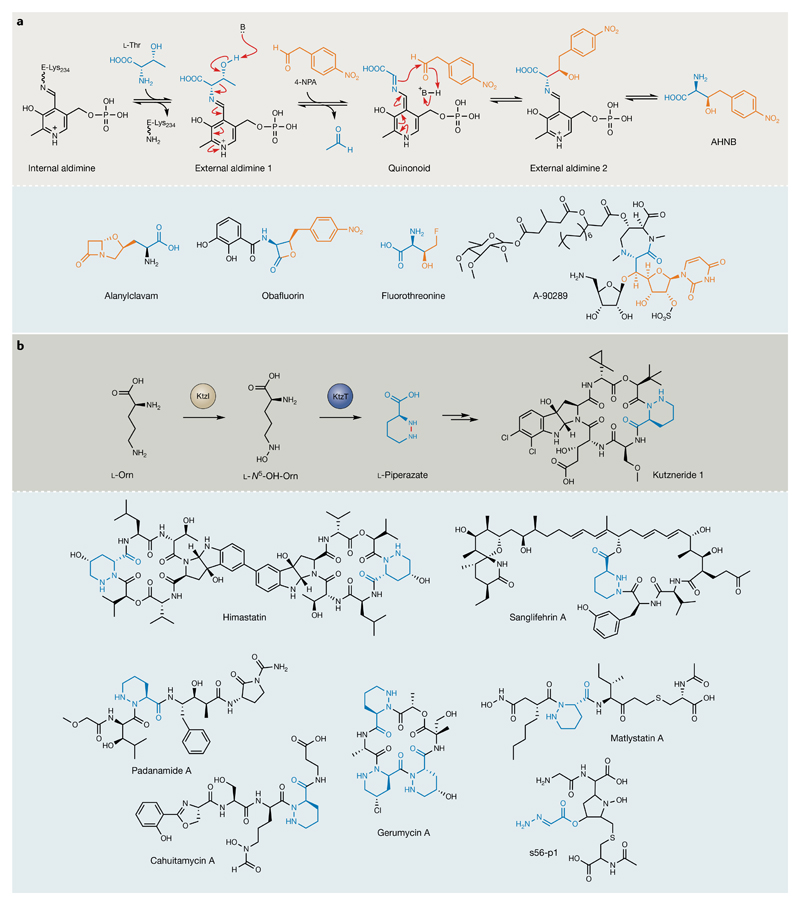
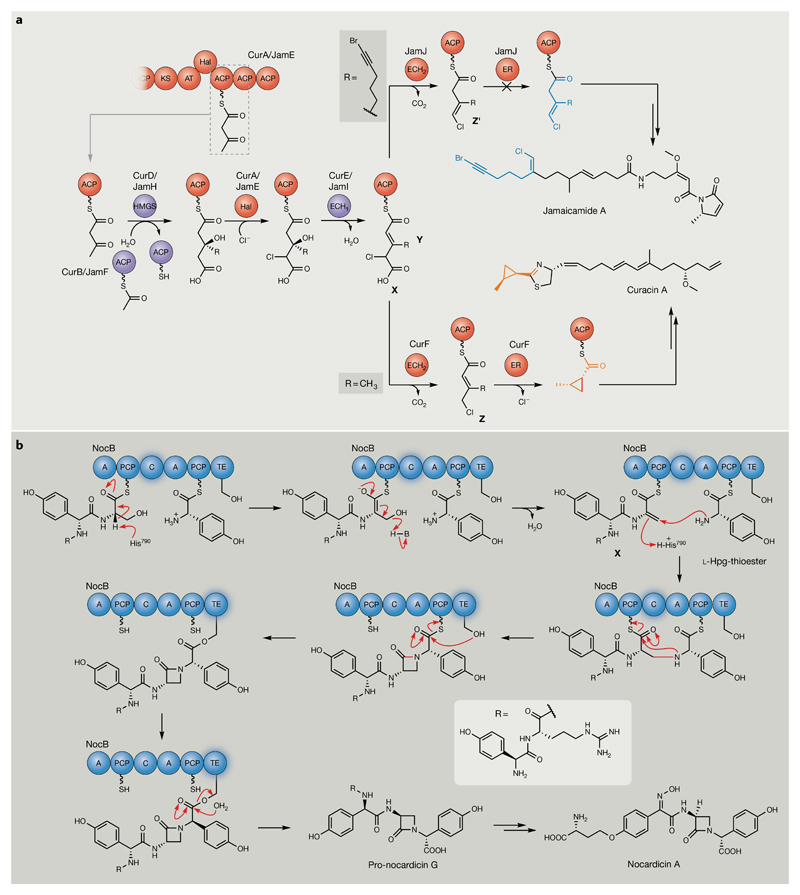
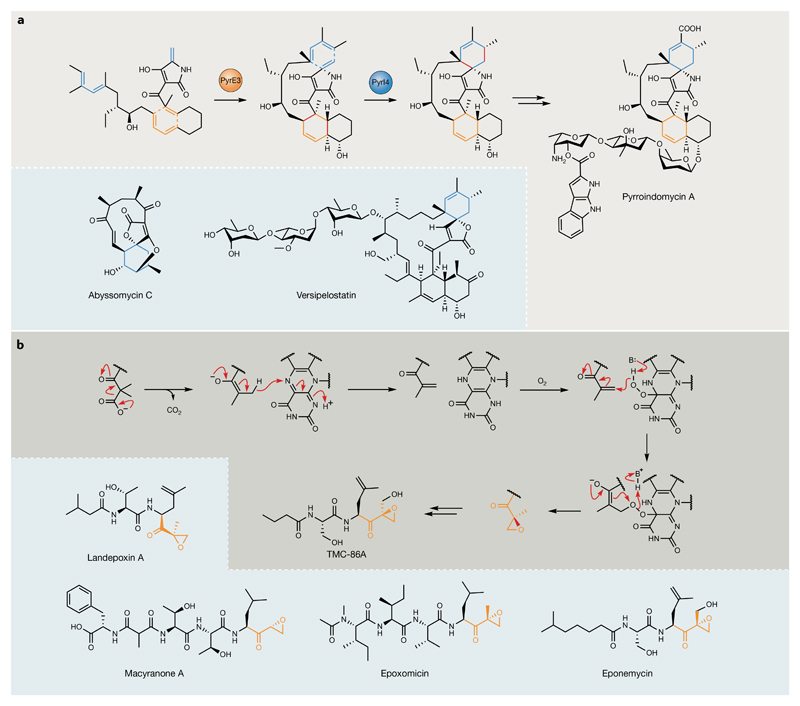
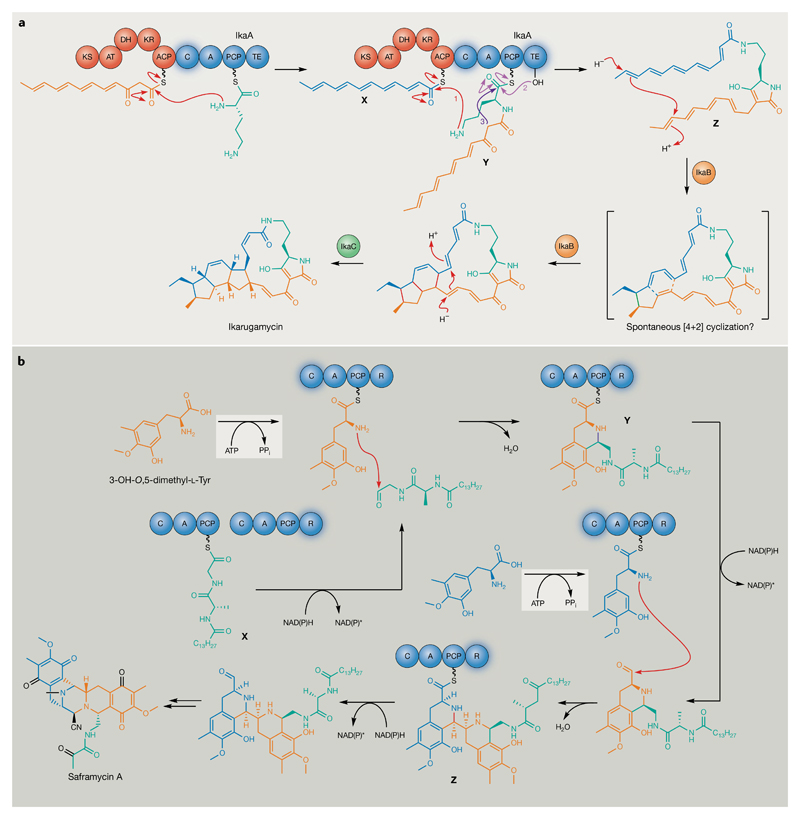
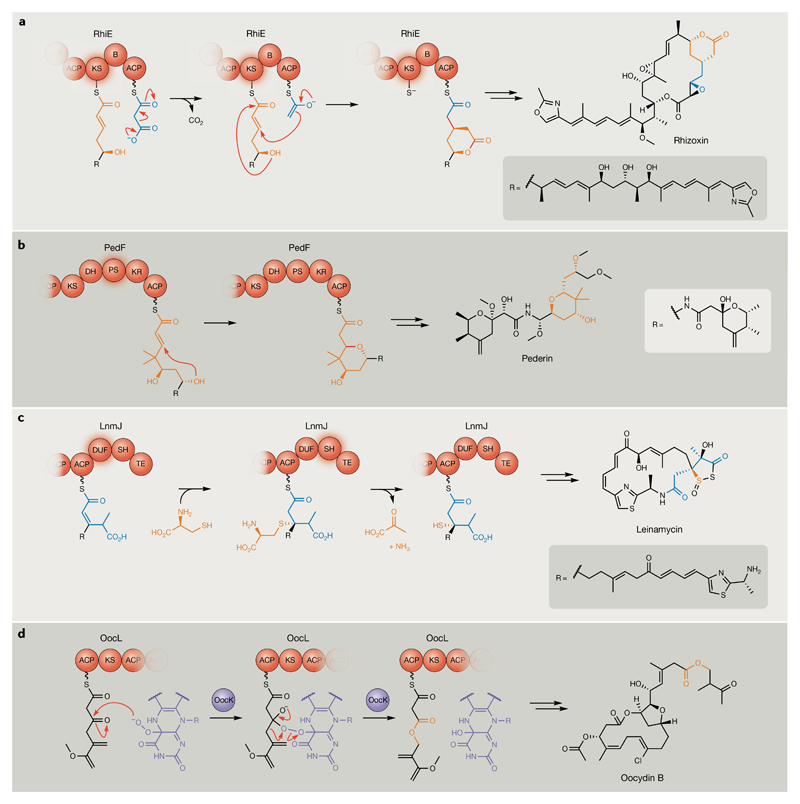
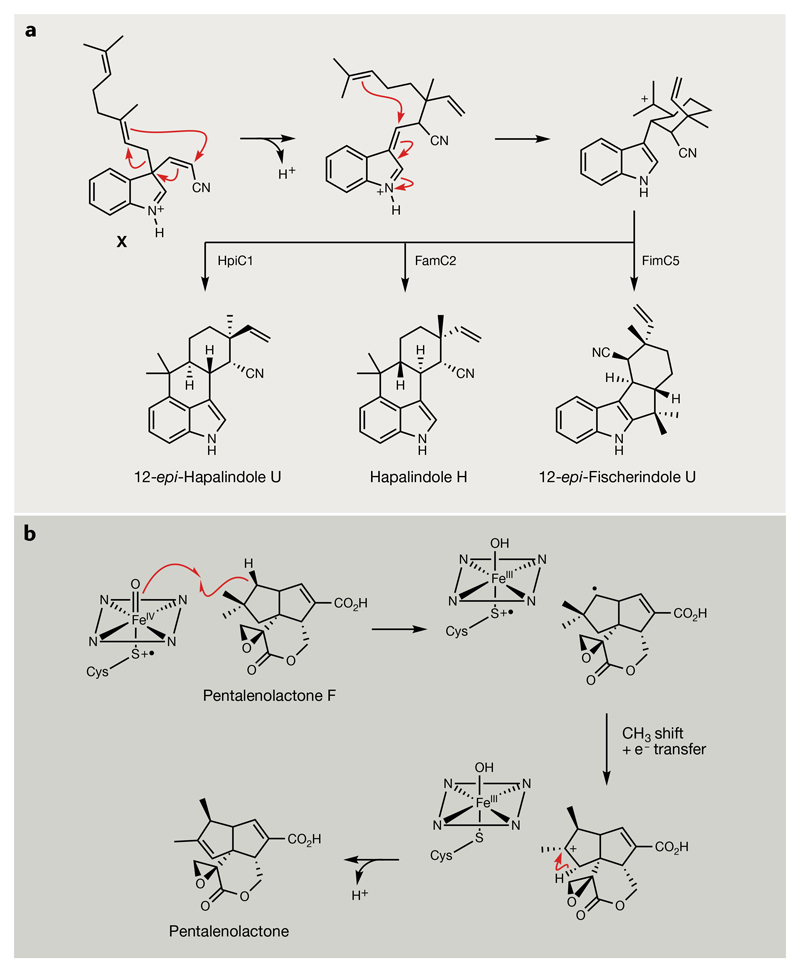
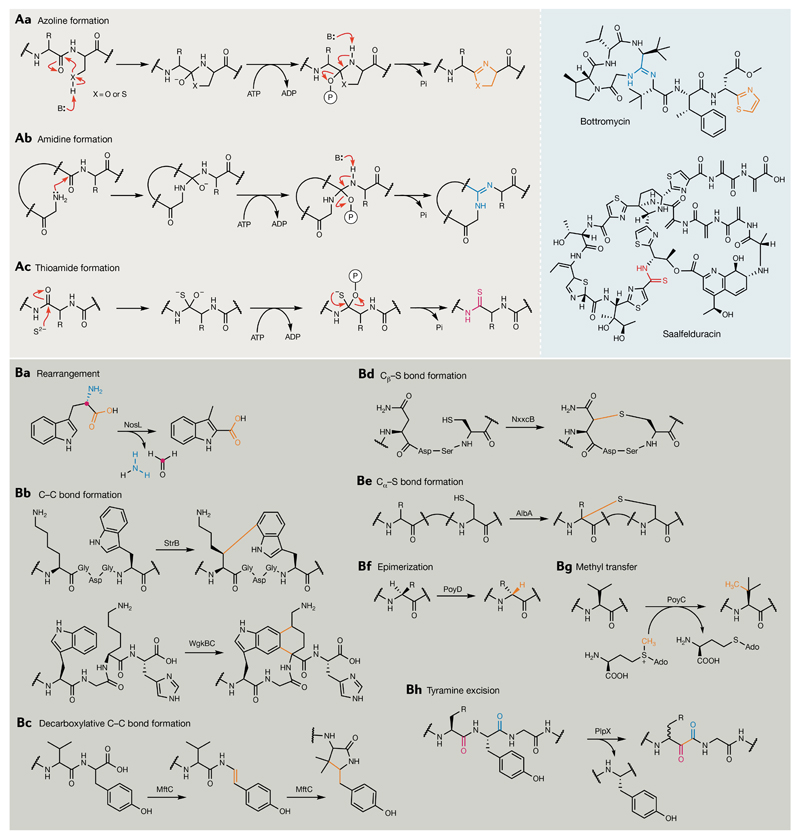
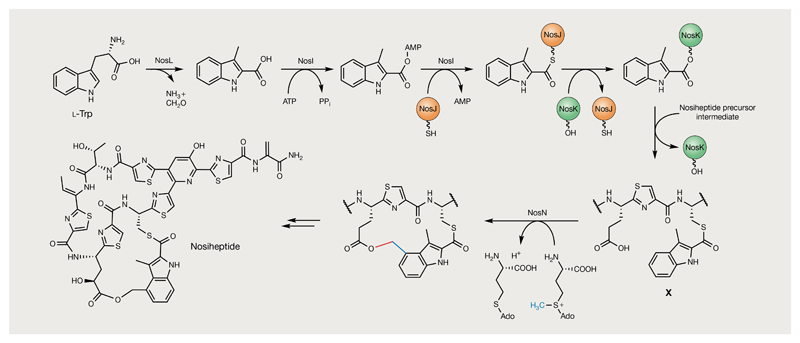
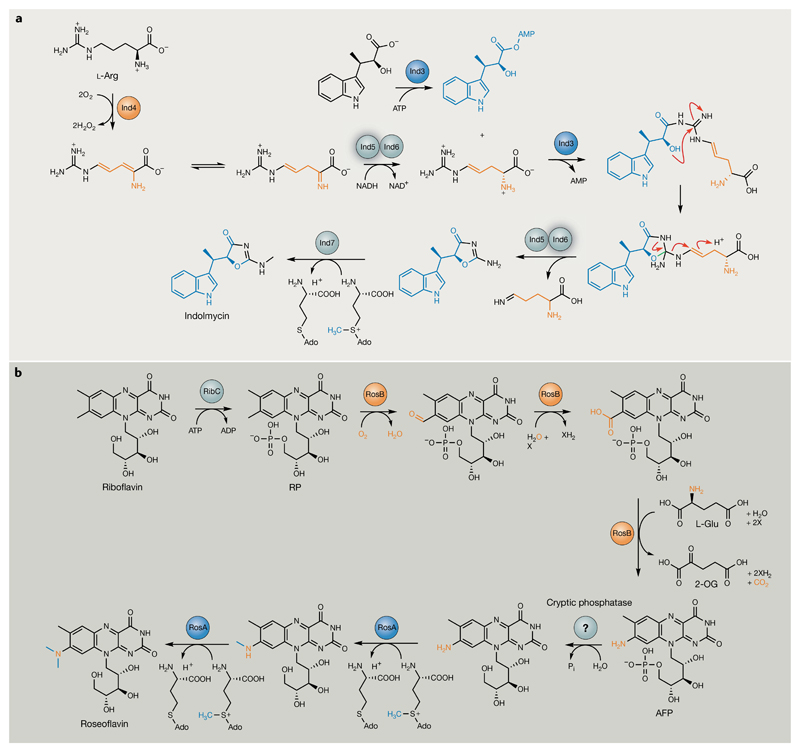
Bacterial natural products display astounding structural diversity, which, in turn, endows them with a remarkable range of biological activities that are of significant value to modern society. Such structural features are generated by biosynthetic enzymes that construct core scaffolds or perform peripheral modifications, and can thus define natural product families, introduce pharmacophores and permit metabolic diversification. Modern genomics approaches have greatly enhanced our ability to access and characterize natural product pathways via sequence-similarity-based bioinformatics discovery strategies. However, many biosynthetic enzymes catalyse exceptional, unprecedented transformations that continue to defy functional prediction and remain hidden from us in bacterial (meta)genomic sequence data. In this Review, we highlight exciting examples of unusual enzymology that have been uncovered recently in the context of natural product biosynthesis. These suggest that much of the natural product diversity, including entire substance classes, awaits discovery. New approaches to lift the veil on the cryptic chemistries of the natural product universe are also discussed.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures

References
-
- Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. - PubMed
-
- Demain AL. Importance of microbial natural products and the need to revitalize their discovery. J Ind Microbiol Biotechnol. 2014;41:185–201. - PubMed
-
- Cantrell CL, Dayan FE, Duke SO. Natural products as sources for new pesticides. J Nat Prod. 2012;75:1231–1242. - PubMed
-
- Oerke E-C, Dehne H-W. Safeguarding production-losses in major crops and the role of crop protection. Crop Prot. 2004;23:275–285.
Grants and funding
LinkOut - more resources
Full Text Sources
