

Deep evolutionary analysis reveals the design principles of fold A glycosyltransferases
- PMID: 32234211
- PMCID: PMC7185993
- DOI: 10.7554/eLife.54532
Deep evolutionary analysis reveals the design principles of fold A glycosyltransferases
Abstract
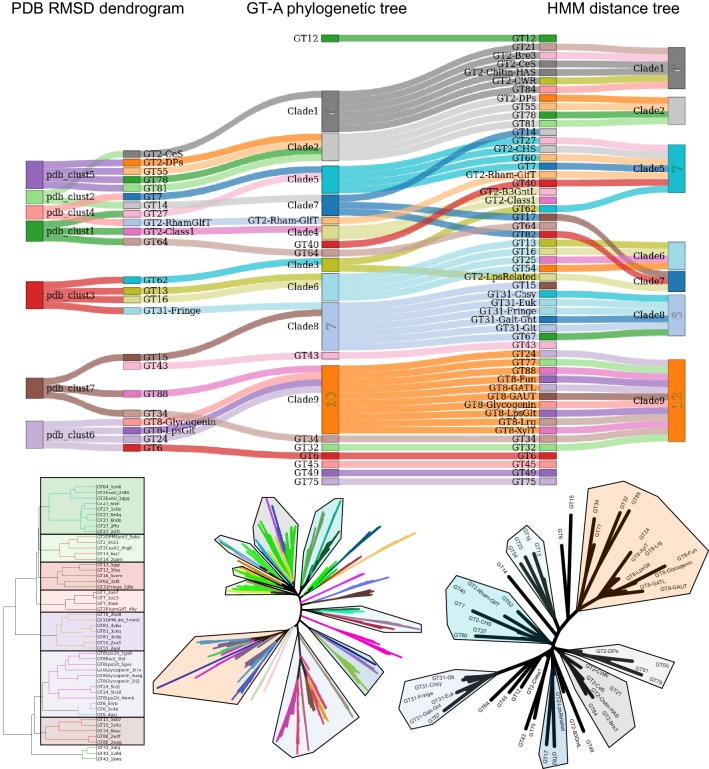
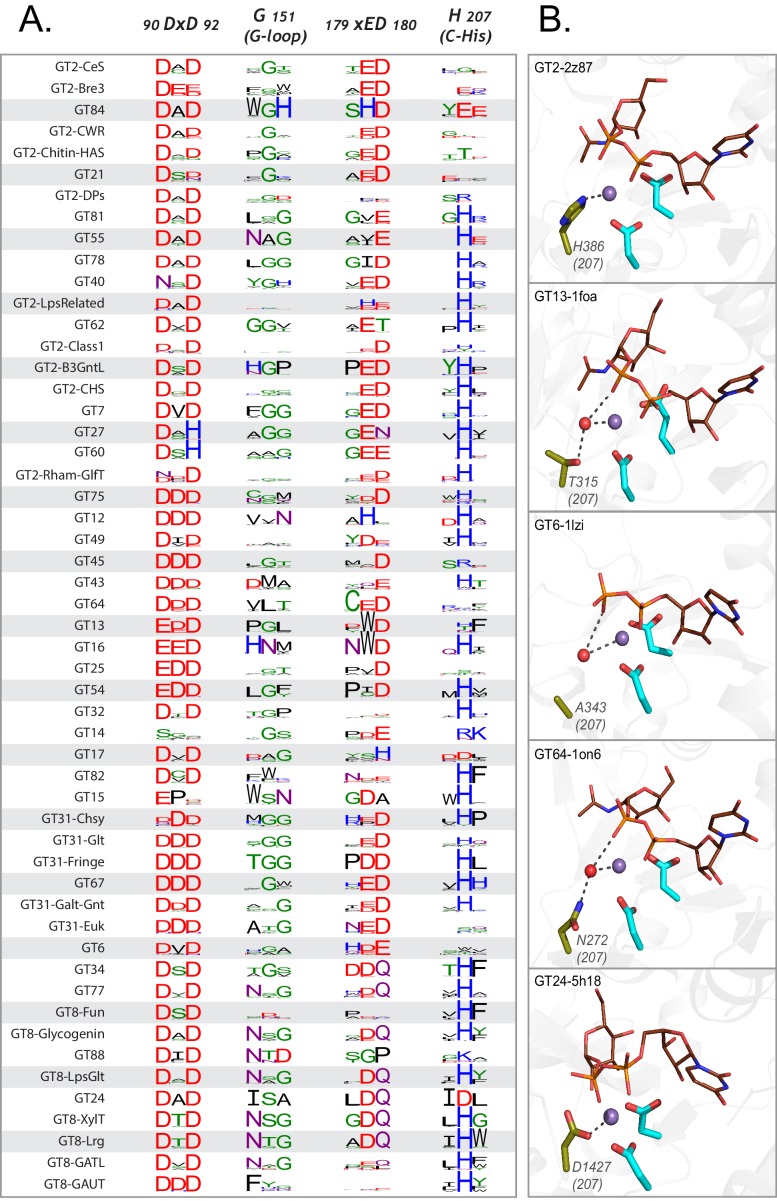
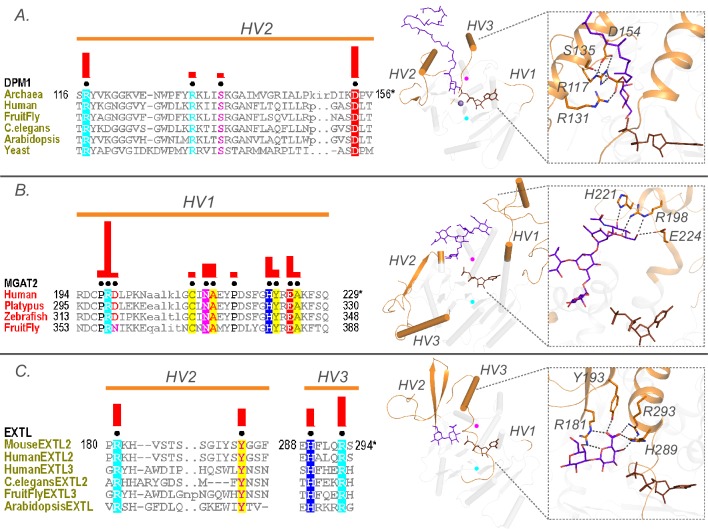
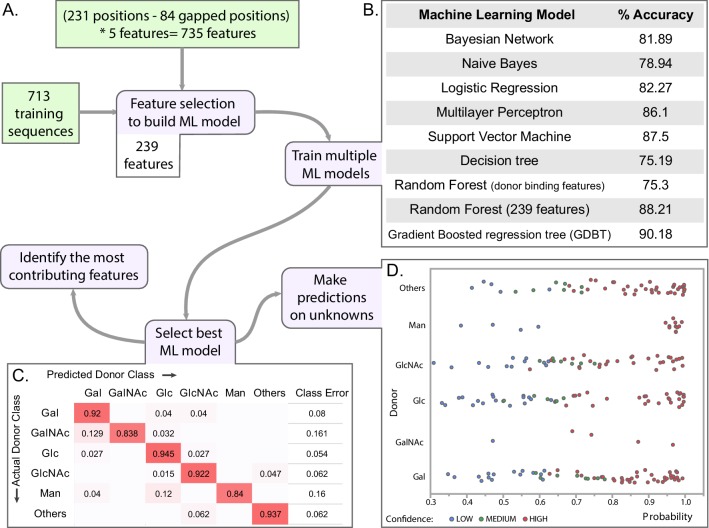
Glycosyltransferases (GTs) are prevalent across the tree of life and regulate nearly all aspects of cellular functions. The evolutionary basis for their complex and diverse modes of catalytic functions remain enigmatic. Here, based on deep mining of over half million GT-A fold sequences, we define a minimal core component shared among functionally diverse enzymes. We find that variations in the common core and emergence of hypervariable loops extending from the core contributed to GT-A diversity. We provide a phylogenetic framework relating diverse GT-A fold families for the first time and show that inverting and retaining mechanisms emerged multiple times independently during evolution. Using evolutionary information encoded in primary sequences, we trained a machine learning classifier to predict donor specificity with nearly 90% accuracy and deployed it for the annotation of understudied GTs. Our studies provide an evolutionary framework for investigating complex relationships connecting GT-A fold sequence, structure, function and regulation.
Keywords: A. thaliana; C. elegans; D. melanogaster; GT evolution; GT phylogeny; S. cerevisiae; common core; computational biology; donor prediction; evolutionary biology; glycosyltransferase; human; machine learning; systems biology.
Plain language summary
Carbohydrates are one of the major groups of large biological molecules that regulate nearly all aspects of life. Yet, unlike DNA or proteins, carbohydrates are made without a template to follow. Instead, these molecules are built from a set of sugar-based building blocks by the intricate activities of a large and diverse family of enzymes known as glycosyltransferases. An incomplete understanding of how glycosyltransferases recognize and build diverse carbohydrates presents a major bottleneck in developing therapeutic strategies for diseases associated with abnormalities in these enzymes. It also limits efforts to engineer these enzymes for biotechnology applications and biofuel production. Taujale et al. have now used evolutionary approaches to map the evolution of a major subset of glycosyltransferases from species across the tree of life to understand how these enzymes evolved such precise mechanisms to build diverse carbohydrates. First, a minimal structural unit was defined based on being shared among a group of over half a million unique glycosyltransferase enzymes with different activities. Further analysis then showed that the diverse activities of these enzymes evolved through the accumulation of mutations within this structural unit, as well as in much more variable regions in the enzyme that extend from the minimal unit. Taujale et al. then built an extended family tree for this collection of glycosyltransferases and details of the evolutionary relationships between the enzymes helped them to create a machine learning framework that could predict which sugar-containing molecules were the raw materials for a given glycosyltransferase. This framework could make predictions with nearly 90% accuracy based only on information that can be deciphered from the gene for that enzyme. These findings will provide scientists with new hypotheses for investigating the complex relationships connecting the genetic information about glycosyltransferases with their structures and activities. Further refinement of the machine learning framework may eventually enable the design of enzymes with properties that are desirable for applications in biotechnology.
© 2020, Taujale et al.
Conflict of interest statement
RT, AV, LH, ZZ, WY, KR, SL, AE, KM, NK No competing interests declared
Figures
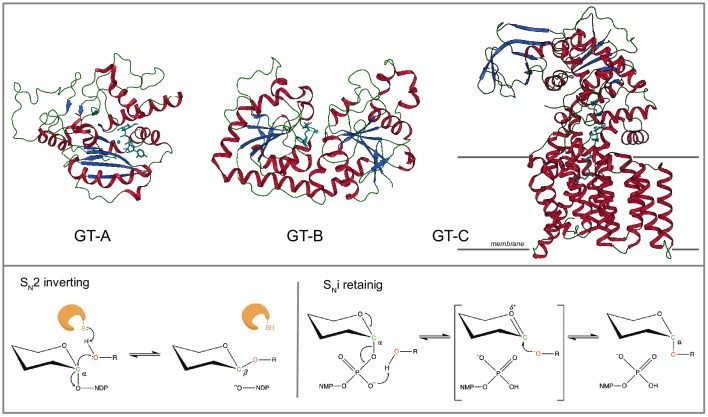
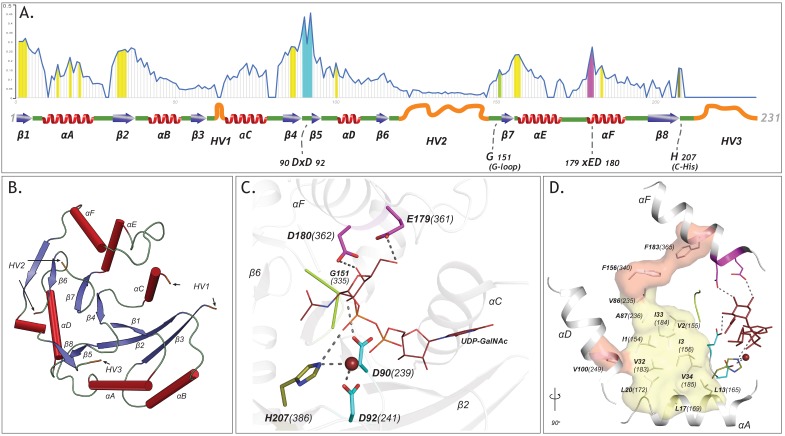
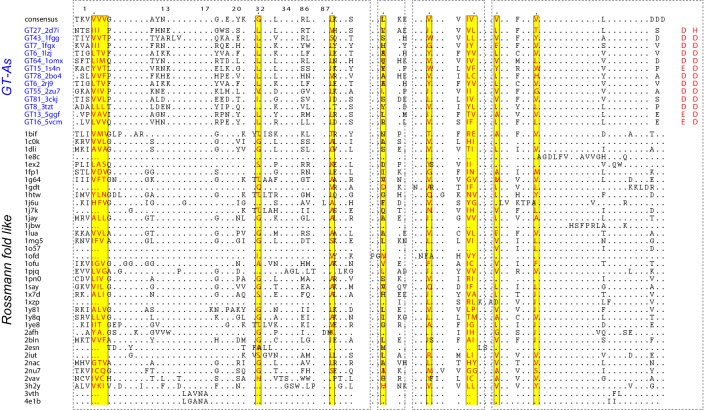
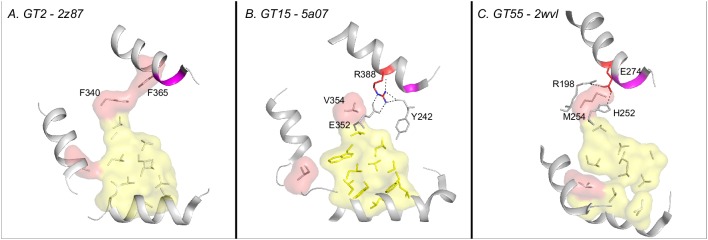
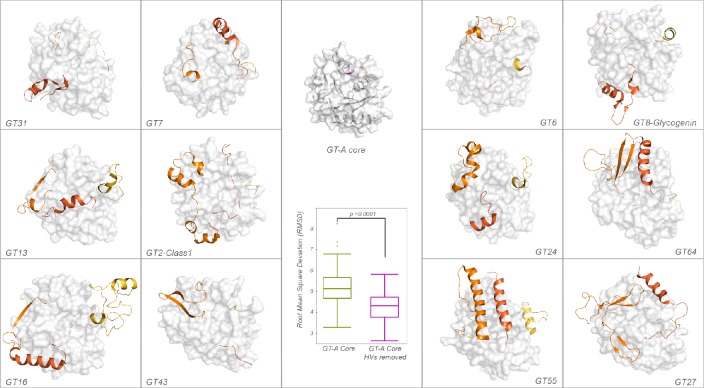
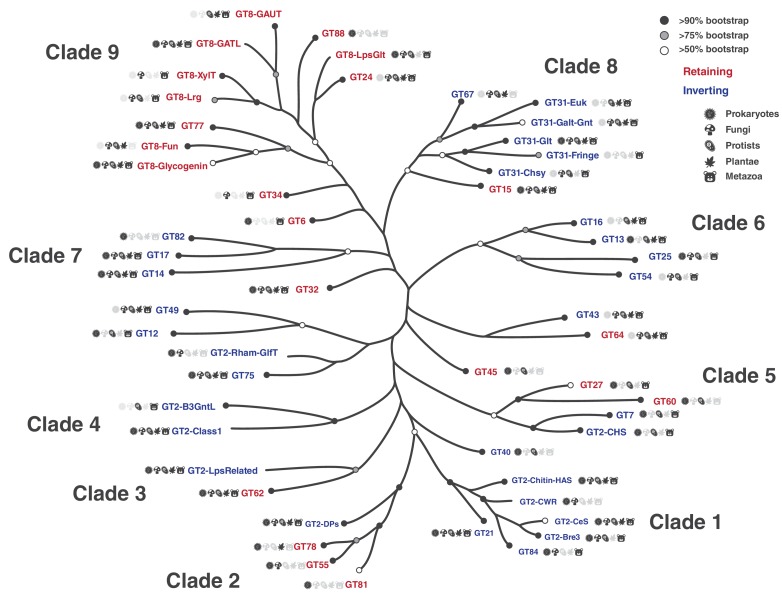
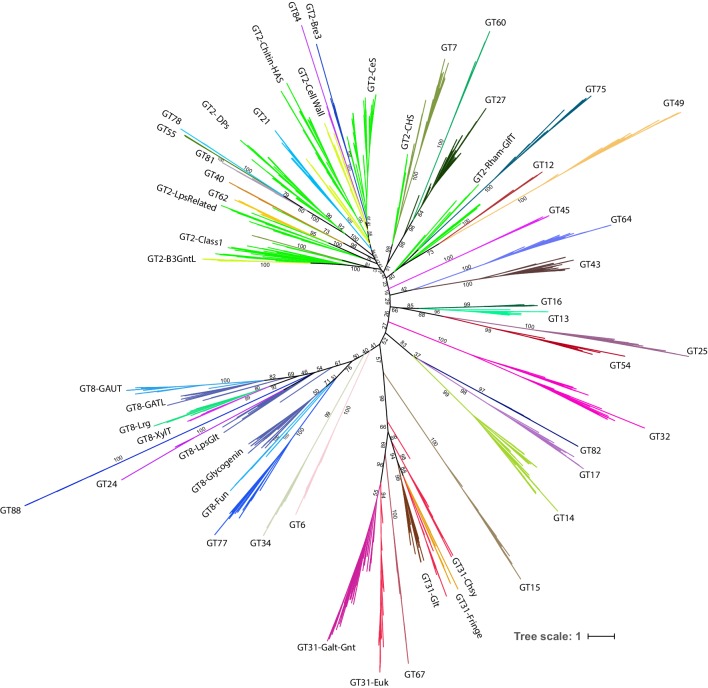
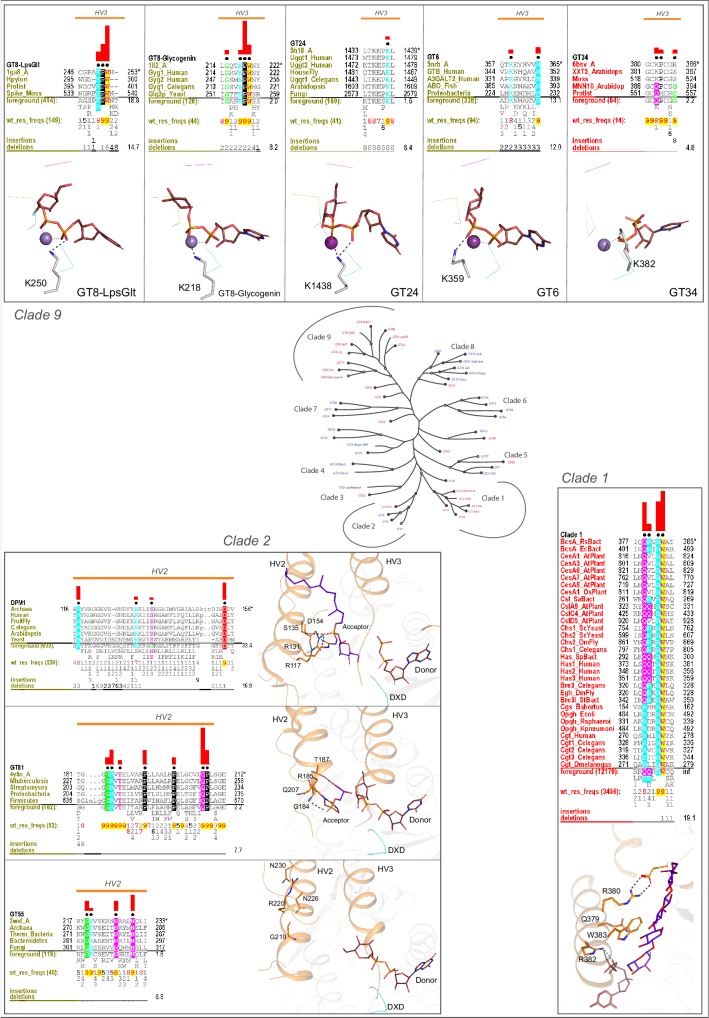
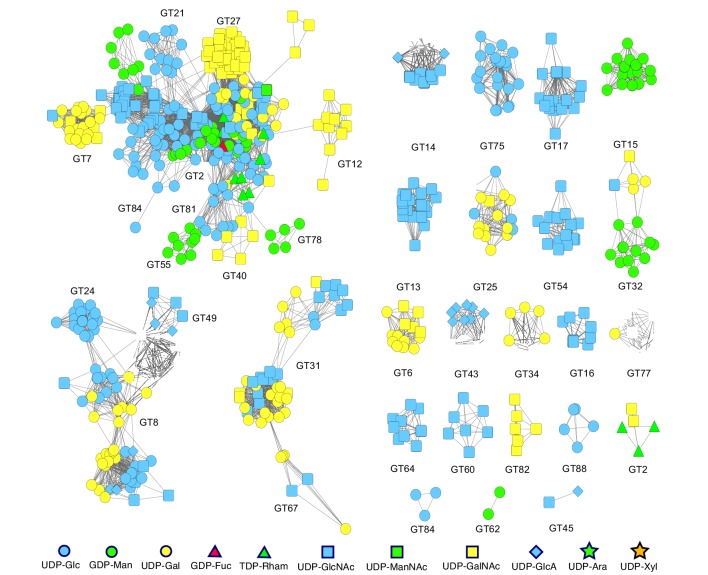
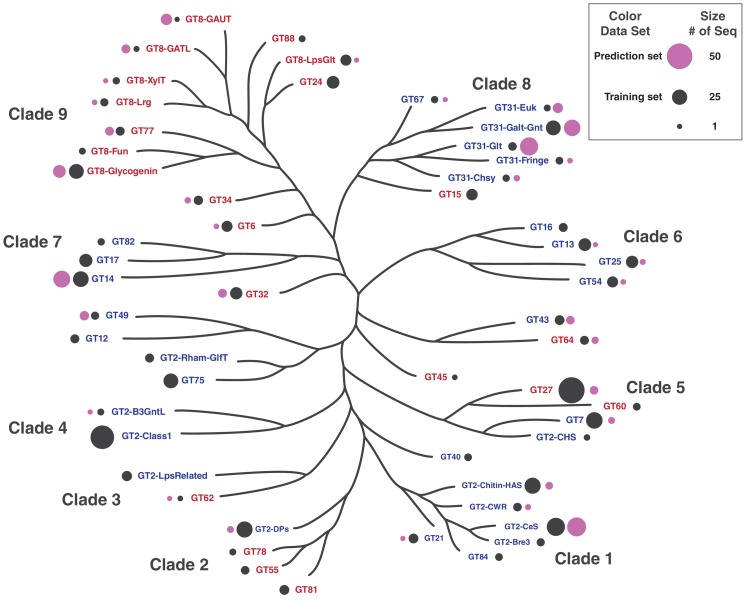
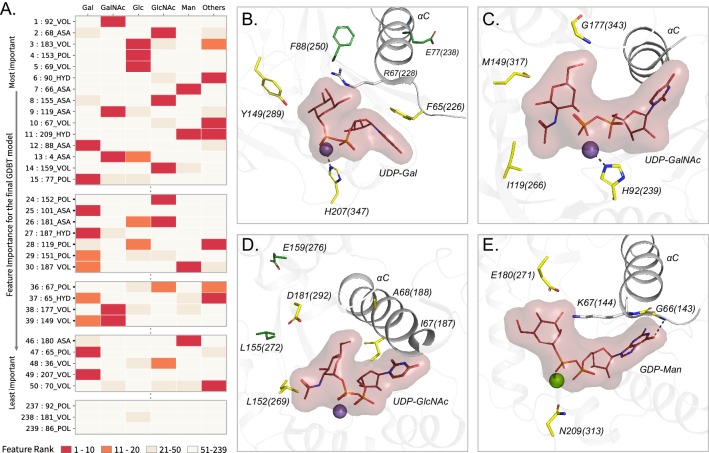
References
-
- Albesa-Jové D, Romero-García J, Sancho-Vaello E, Contreras FX, Rodrigo-Unzueta A, Comino N, Carreras-González A, Arrasate P, Urresti S, Biarnés X, Planas A, Guerin ME. Structural snapshots and loop dynamics along the catalytic cycle of glycosyltransferase GpgS. Structure. 2017;25:1034–1044. doi: 10.1016/j.str.2017.05.009. - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
