

Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies
- PMID: 32234780
- PMCID: PMC7165463
- DOI: 10.1073/pnas.1919960117
Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies
Abstract
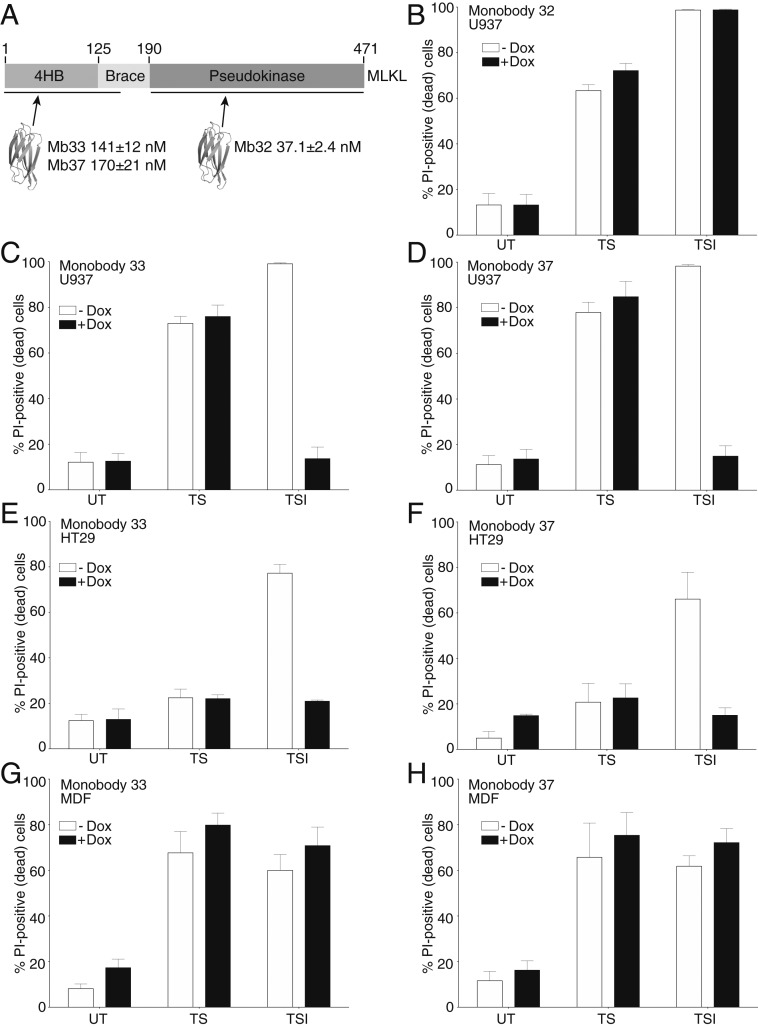
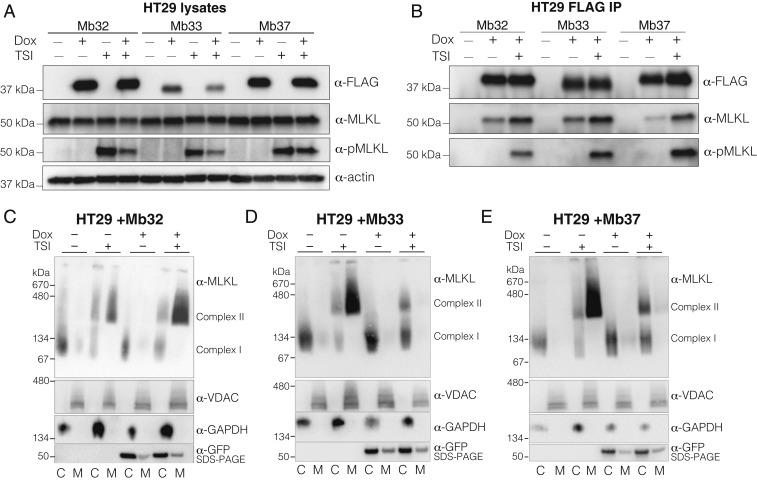
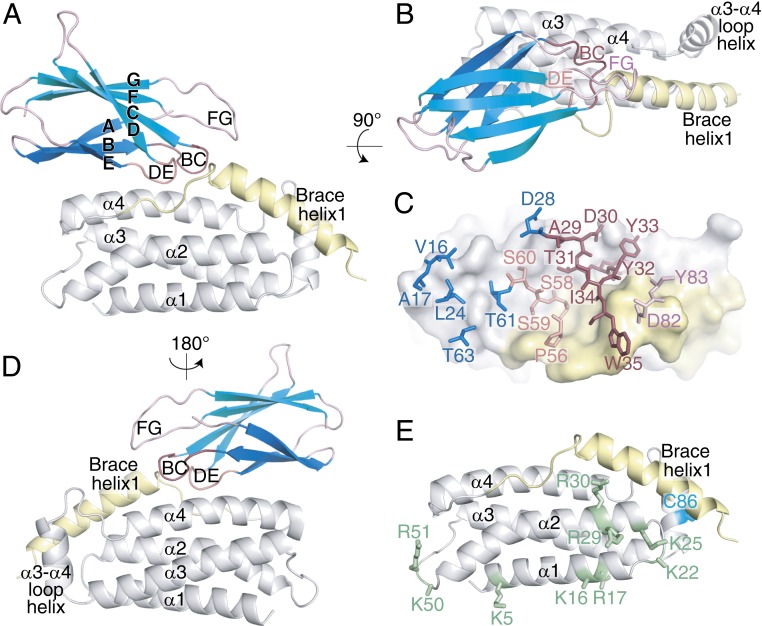
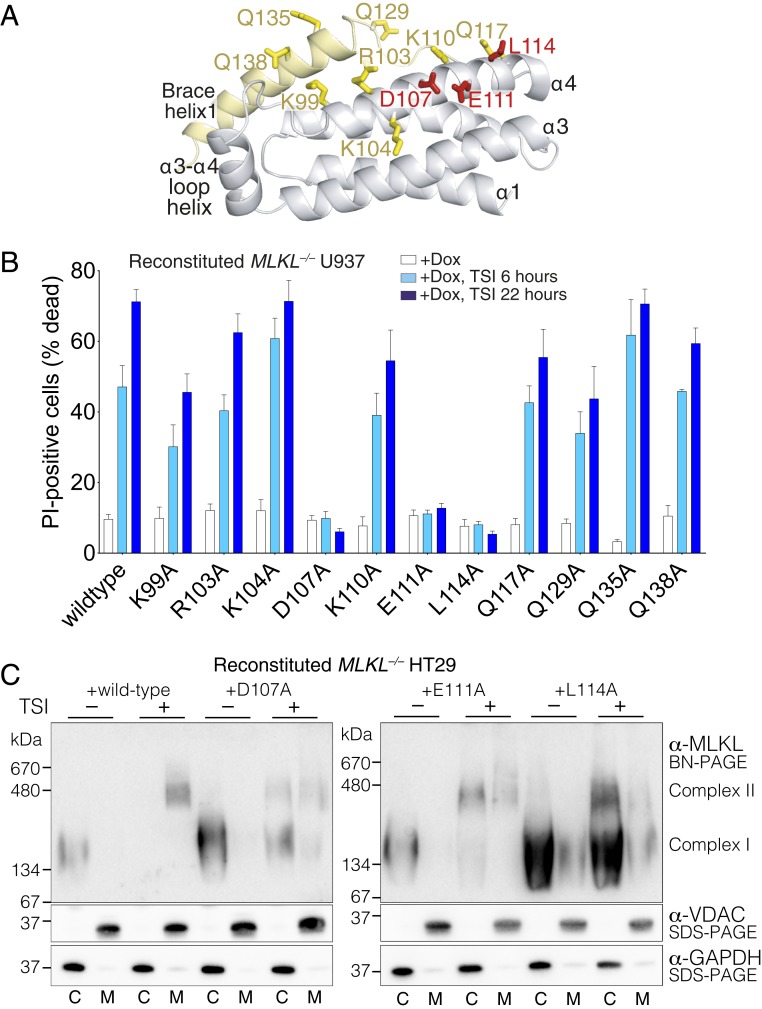
The necroptosis cell death pathway has been implicated in host defense and in the pathology of inflammatory diseases. While phosphorylation of the necroptotic effector pseudokinase Mixed Lineage Kinase Domain-Like (MLKL) by the upstream protein kinase RIPK3 is a hallmark of pathway activation, the precise checkpoints in necroptosis signaling are still unclear. Here we have developed monobodies, synthetic binding proteins, that bind the N-terminal four-helix bundle (4HB) "killer" domain and neighboring first brace helix of human MLKL with nanomolar affinity. When expressed as genetically encoded reagents in cells, these monobodies potently block necroptotic cell death. However, they did not prevent MLKL recruitment to the "necrosome" and phosphorylation by RIPK3, nor the assembly of MLKL into oligomers, but did block MLKL translocation to membranes where activated MLKL normally disrupts membranes to kill cells. An X-ray crystal structure revealed a monobody-binding site centered on the α4 helix of the MLKL 4HB domain, which mutational analyses showed was crucial for reconstitution of necroptosis signaling. These data implicate the α4 helix of its 4HB domain as a crucial site for recruitment of adaptor proteins that mediate membrane translocation, distinct from known phospholipid binding sites.
Keywords: RIPK3; cell death; programmed necrosis; protein engineering; protein interactions.
Conflict of interest statement
Competing interest statement: E.J.P., J.M.H., S.E.G., A.L.S., C.F., S.N.Y., P.E.C., and J.M.M. contribute to a project developing necroptosis inhibitors with Anaxis Pharma Pty Ltd. A.K. and S.K. are listed as inventors on issued and pending patents on the monobody technology filed by The University of Chicago (US Patent 9512199 B2 and related pending applications).
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Miscellaneous
