

Selection of Features with Consistent Profiles Improves Relative Protein Quantification in Mass Spectrometry Experiments
- PMID: 32234965
- PMCID: PMC7261813
- DOI: 10.1074/mcp.RA119.001792
Selection of Features with Consistent Profiles Improves Relative Protein Quantification in Mass Spectrometry Experiments
Abstract
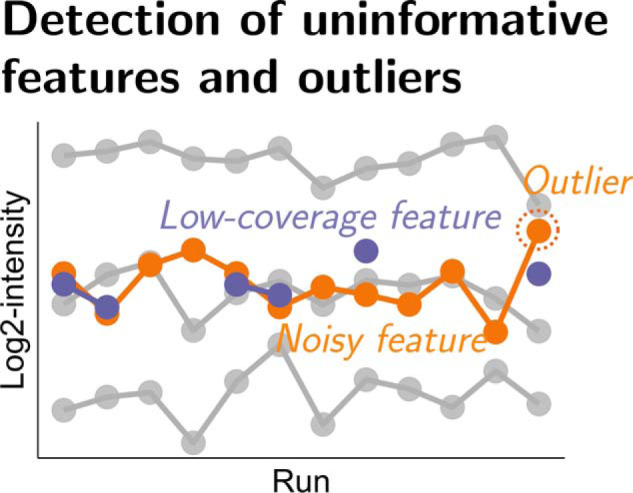
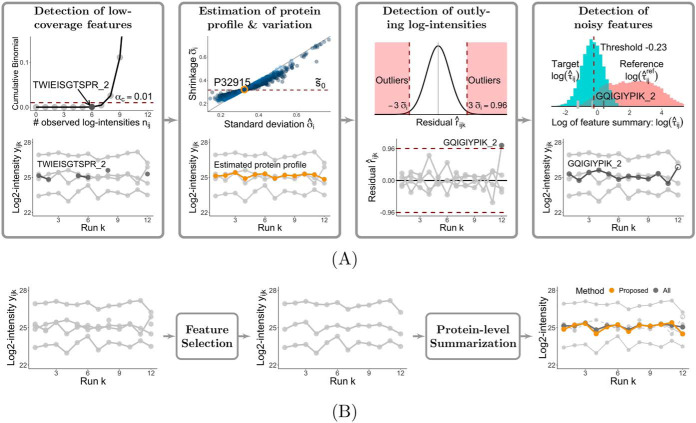
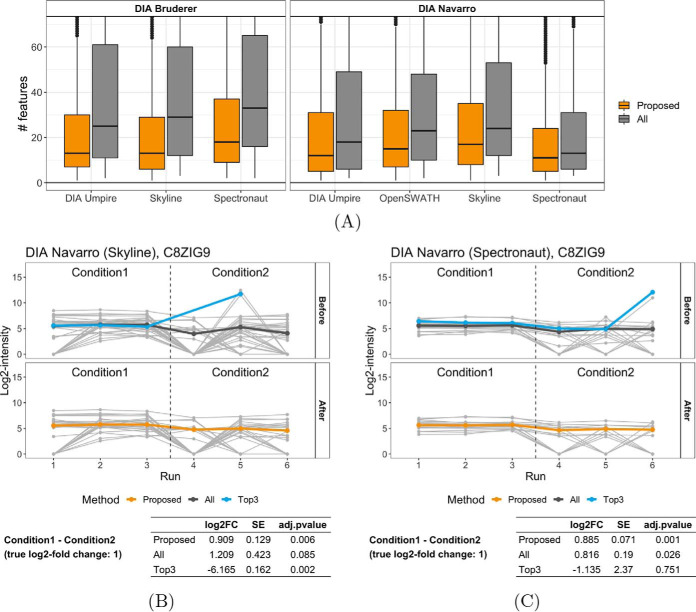
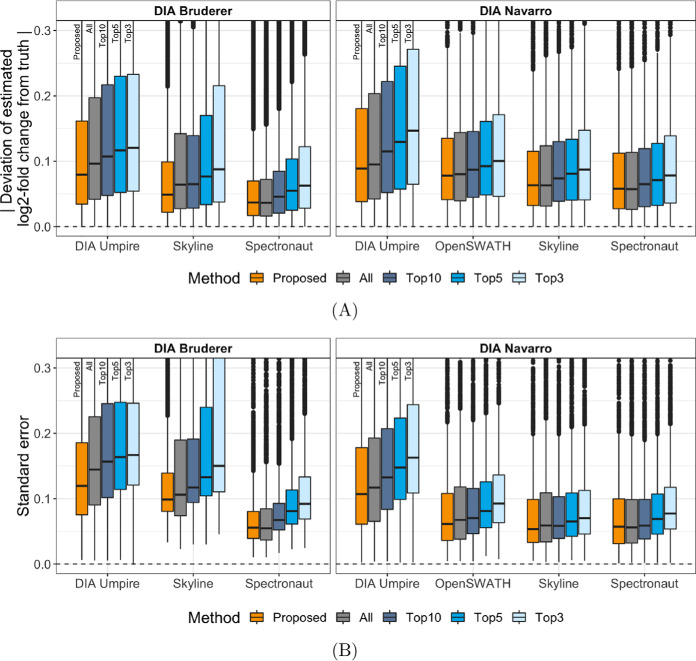
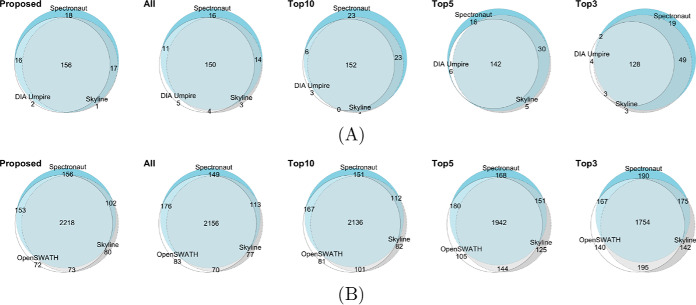
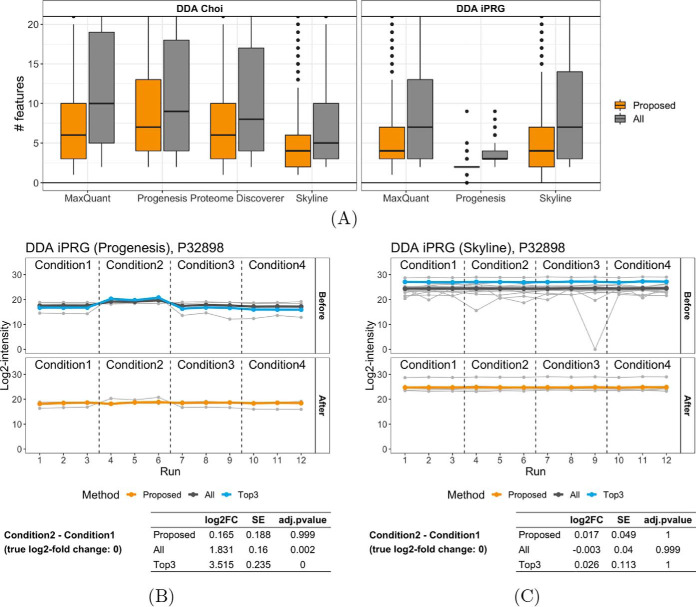
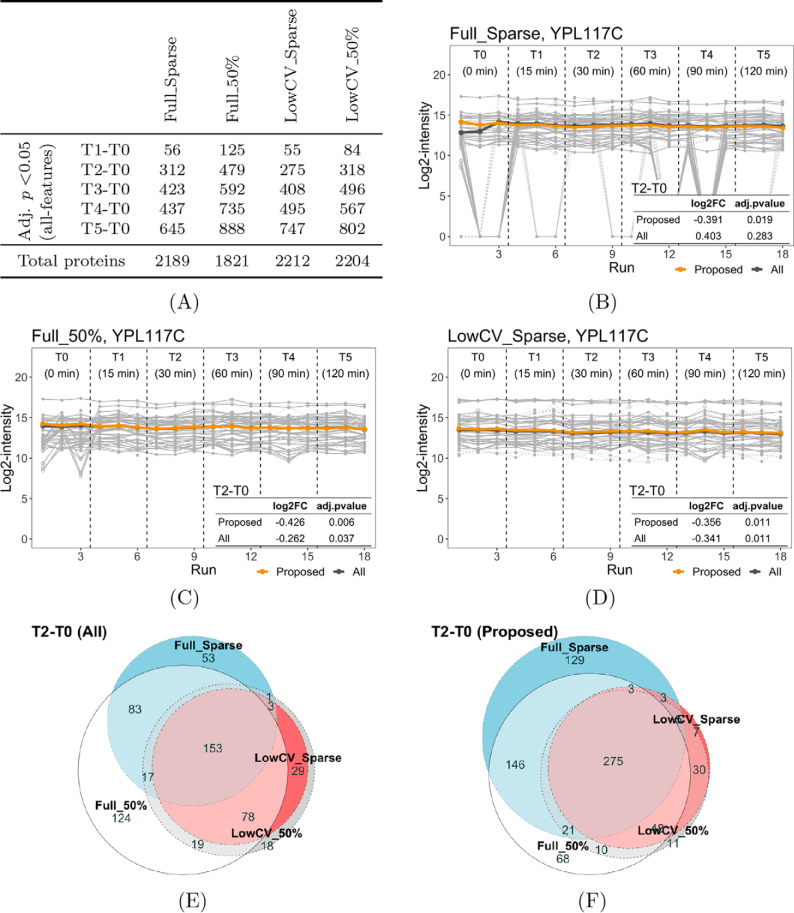
In bottom-up mass spectrometry-based proteomics, relative protein quantification is often achieved with data-dependent acquisition (DDA), data-independent acquisition (DIA), or selected reaction monitoring (SRM). These workflows quantify proteins by summarizing the abundances of all the spectral features of the protein (e.g. precursor ions, transitions or fragments) in a single value per protein per run. When abundances of some features are inconsistent with the overall protein profile (for technological reasons such as interferences, or for biological reasons such as post-translational modifications), the protein-level summaries and the downstream conclusions are undermined. We propose a statistical approach that automatically detects spectral features with such inconsistent patterns. The detected features can be separately investigated, and if necessary, removed from the data set. We evaluated the proposed approach on a series of benchmark-controlled mixtures and biological investigations with DDA, DIA and SRM data acquisitions. The results demonstrated that it could facilitate and complement manual curation of the data. Moreover, it can improve the estimation accuracy, sensitivity and specificity of detecting differentially abundant proteins, and reproducibility of conclusions across different data processing tools. The approach is implemented as an option in the open-source R-based software MSstats.
Keywords: Statistics; bioinformatics; biostatistics; computational biology; label-free quantification; mass spectrometry; multiple reaction monitoring; quantification; selected reaction monitoring; targeted mass spectrometry.
© 2020 Tsai et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Aebersold R., and Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 - PubMed
-
- Aebersold R., and Mann M. (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 - PubMed
-
- Bruderer R., Bernhardt O. M., Gandhi T., Miladinović S. M., Cheng L.-Y., Messner S., Ehrenberger T., Zanotelli V., Butscheid Y., Escher C., Vitek O., Rinner O., and Reiter L. (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410 - PMC - PubMed
-
- Deleted in proof.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
