

Diagnosis of Mucopolysaccharidoses
- PMID: 32235807
- PMCID: PMC7151013
- DOI: 10.3390/diagnostics10030172
Diagnosis of Mucopolysaccharidoses
Abstract
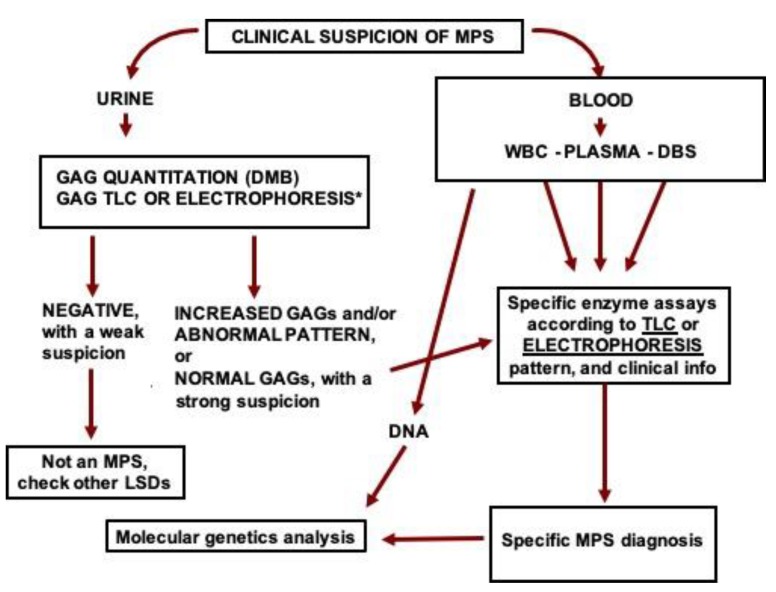
The mucopolysaccharidoses (MPSs) include 11 different conditions caused by specific enzyme deficiencies in the degradation pathway of glycosaminoglycans (GAGs). Although most MPS types present increased levels of GAGs in tissues, including blood and urine, diagnosis is challenging as specific enzyme assays are needed for the correct diagnosis. Enzyme assays are usually performed in blood, with some samples (as leukocytes) providing a final diagnosis, while others (such as dried blood spots) still being considered as screening methods. The identification of variants in the specific genes that encode each MPS-related enzyme is helpful for diagnosis confirmation (when needed), carrier detection, genetic counseling, prenatal diagnosis (preferably in combination with enzyme assays) and phenotype prediction. Although the usual diagnostic flow in high-risk patients starts with the measurement of urinary GAGs, it continues with specific enzyme assays and is completed with mutation identification; there is a growing trend to have genotype-based investigations performed at the beginning of the investigation. In such cases, confirmation of pathogenicity of the variants identified should be confirmed by measurement of enzyme activity and/or identification and/or quantification of GAG species. As there is a growing number of countries performing newborn screening for MPS diseases, the investigation of a low enzyme activity by the measurement of GAG species concentration and identification of gene mutations in the same DBS sample is recommended before the suspicion of MPS is taken to the family. With specific therapies already available for most MPS patients, and with clinical trials in progress for many conditions, the specific diagnosis of MPS as early as possible is becoming increasingly necessary. In this review, we describe traditional and the most up to date diagnostic methods for mucopolysaccharidoses.
Keywords: enzyme replacement therapy; glycosaminoglycans; mucopolysaccharidoses; newborn screening.; tandem mass spectrometry.
Conflict of interest statement
R.G. has received in the last 12 months speaker fees, expert honorarium, travel and/or research grants from Abeona, Actelion, Amicus, Armagen, BioMarin, CentoGene, G.C. Pharma, Inventiva, J.C.R. Pharmaceuticals, Lysogene, Protalix, P.T.C., RegenxBio, Sanofi Genzyme, Shire, Sobi and Ultragenyx. The other authors declare no conflict of interest.
Figures
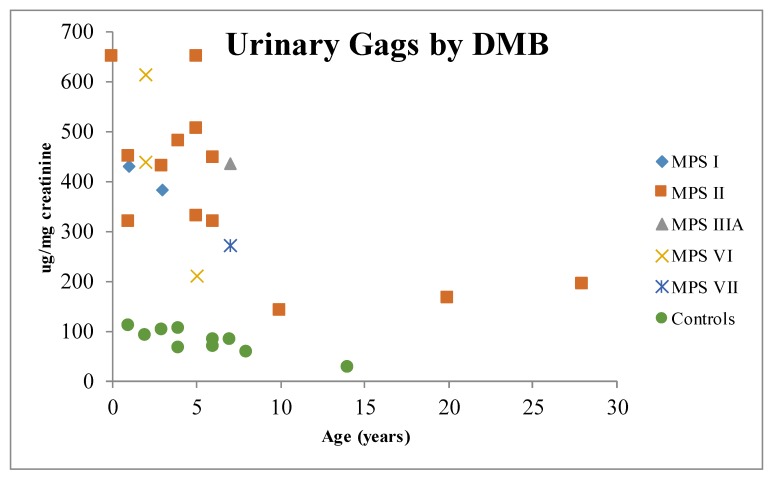
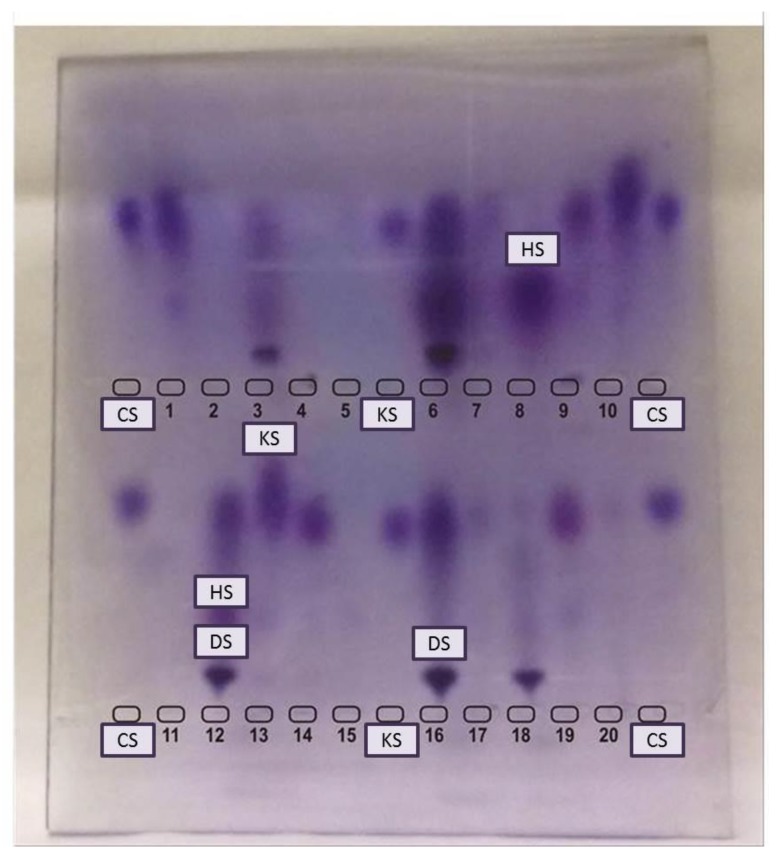
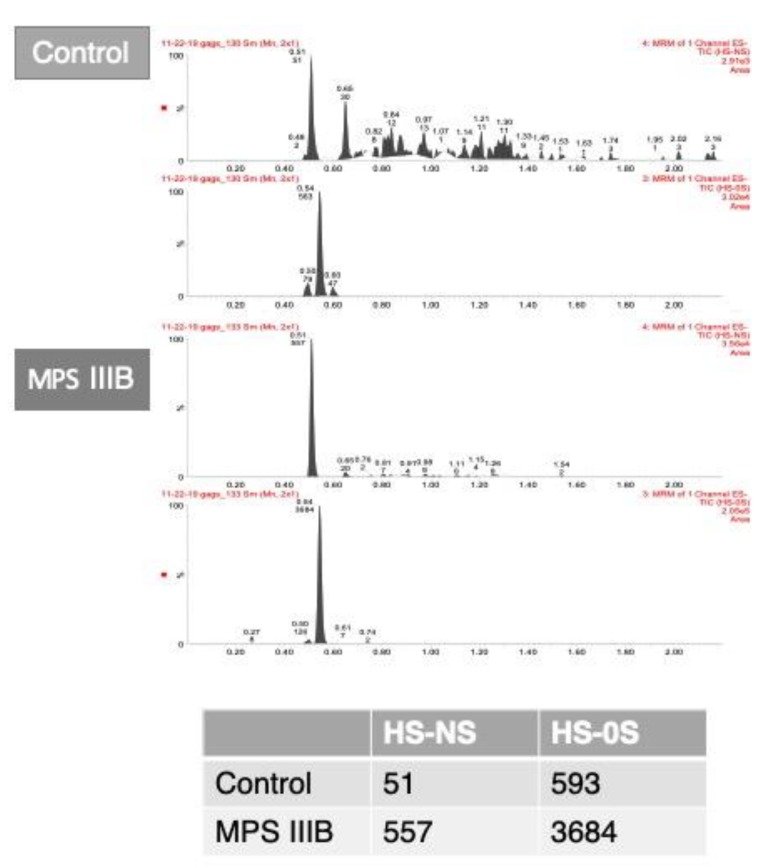
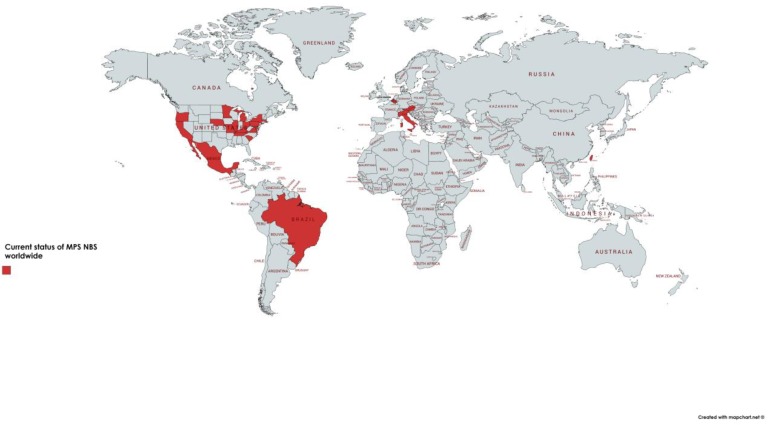
References
-
- Neufeld E., Muenzer J. The Mucopolysaccharidoses | The Online Metabolic and Molecular Bases of Inherited Disease | OMMBID | McGraw-Hill Medical. 8th ed. McGraw-Hill; New York, NY, USA: 2001.
-
- Civallero G., Michelin K., de Mari J., Viapiana M., Burin M., Coelho J.C., Giugliani R. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin. Chim. Acta. 2006;372:98–102. doi: 10.1016/j.cca.2006.03.029. - DOI - PubMed
Publication types
Grants and funding
- 465549/2014-4/Conselho Nacional de Desenvolvimento Científico e Tecnológico
- 405495/2018-8/Conselho Nacional de Desenvolvimento Científico e Tecnológico
- 88887.136366/2017-00/Coordenação de Aperfeiçoamento de Pessoal de Nível Superior
- 17/2551-0000521-0/Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul
- 17-0445/FIPE-HCPA
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
