

Lineage tracing meets single-cell omics: opportunities and challenges
- PMID: 32235876
- PMCID: PMC7307462
- DOI: 10.1038/s41576-020-0223-2
Lineage tracing meets single-cell omics: opportunities and challenges
Abstract
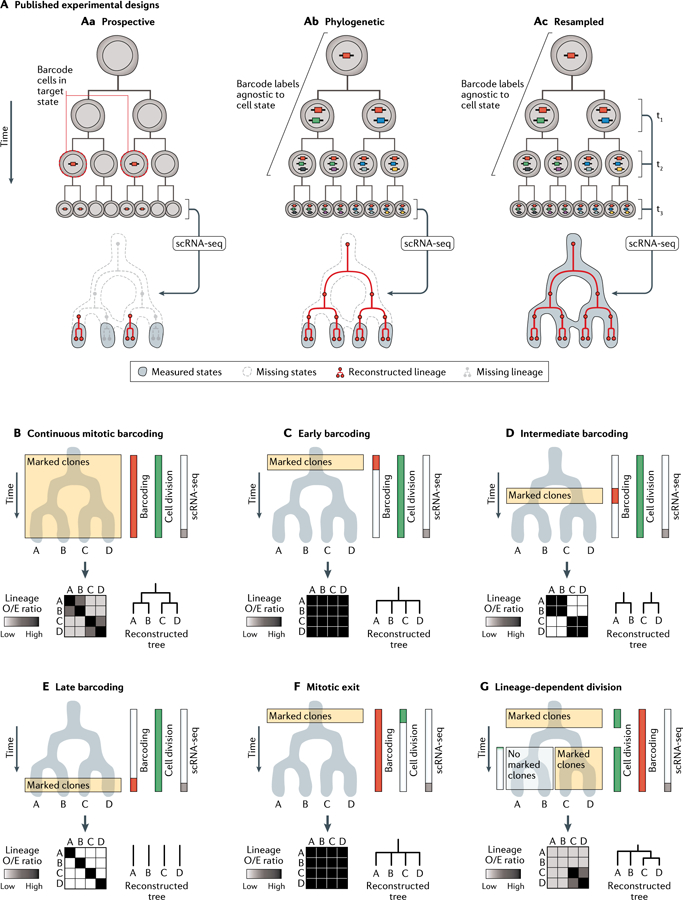
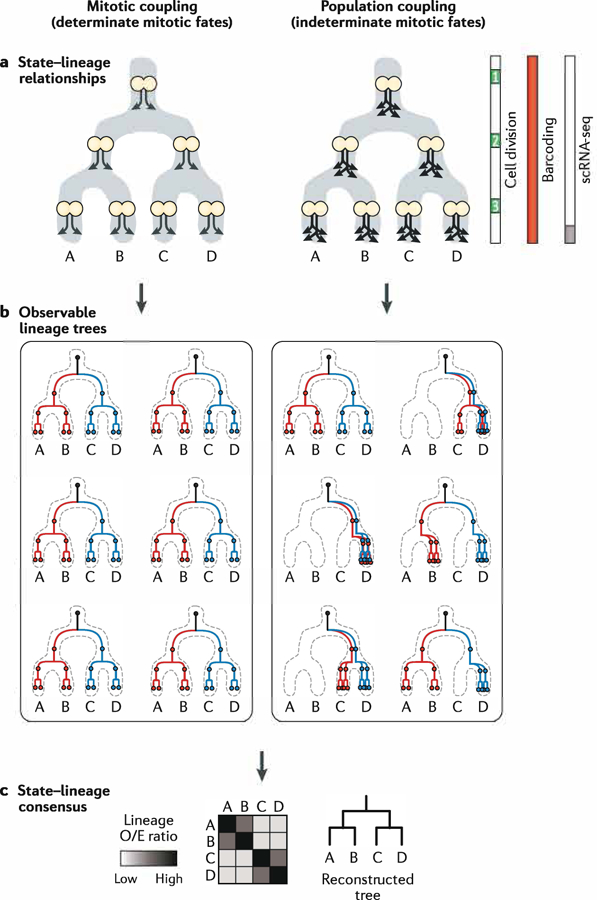
A fundamental goal of developmental and stem cell biology is to map the developmental history (ontogeny) of differentiated cell types. Recent advances in high-throughput single-cell sequencing technologies have enabled the construction of comprehensive transcriptional atlases of adult tissues and of developing embryos from measurements of up to millions of individual cells. Parallel advances in sequencing-based lineage-tracing methods now facilitate the mapping of clonal relationships onto these landscapes and enable detailed comparisons between molecular and mitotic histories. Here we review recent progress and challenges, as well as the opportunities that emerge when these two complementary representations of cellular history are synthesized into integrated models of cell differentiation.
Conflict of interest statement
Competing interests
A.M.K. is a founder of 1CellBio, Inc. D.E.W. declares no competing interests.
Figures
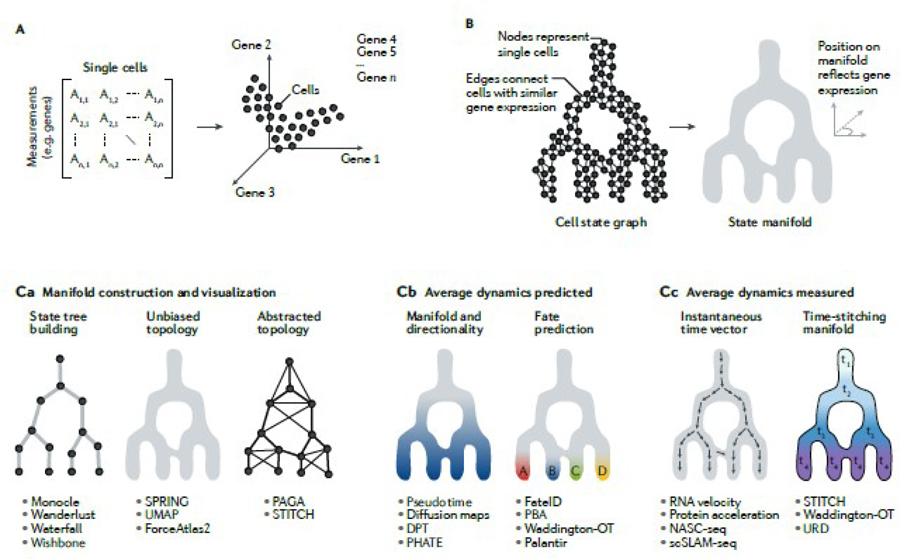
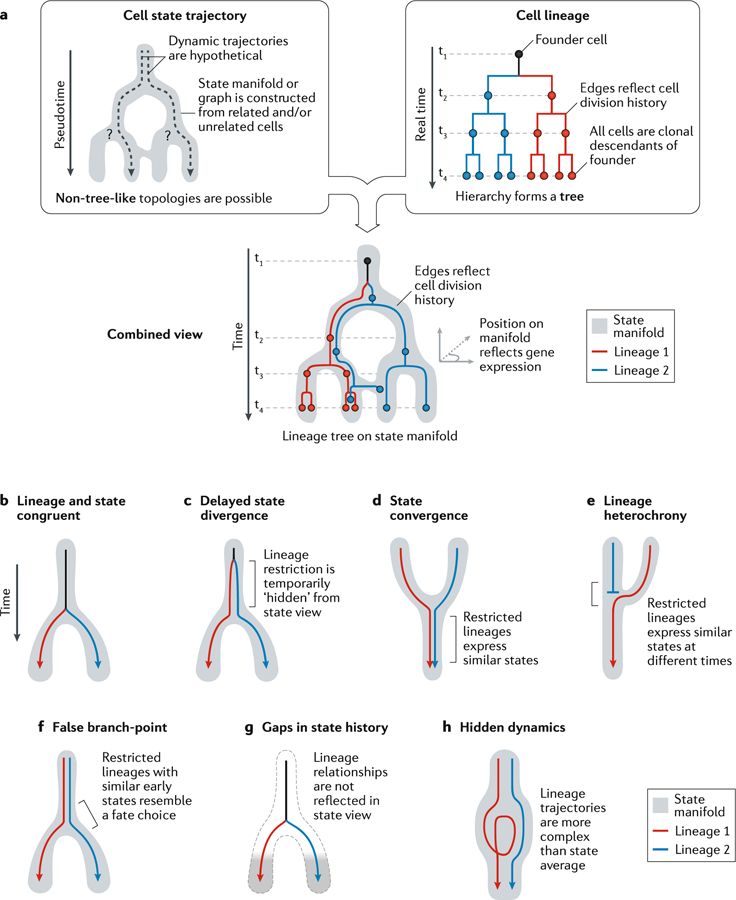
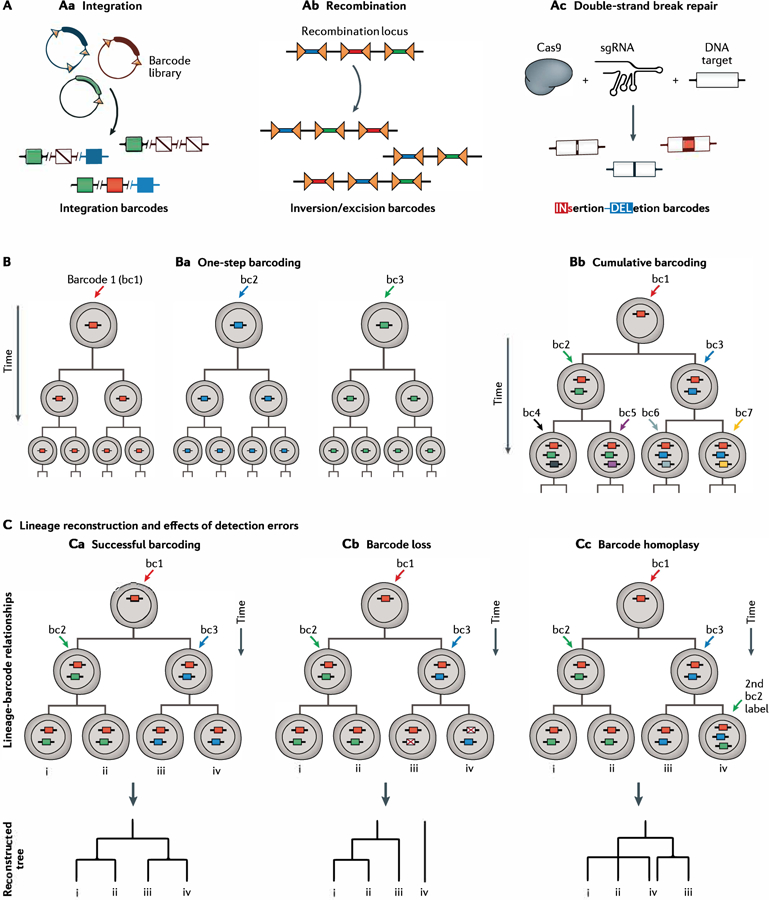
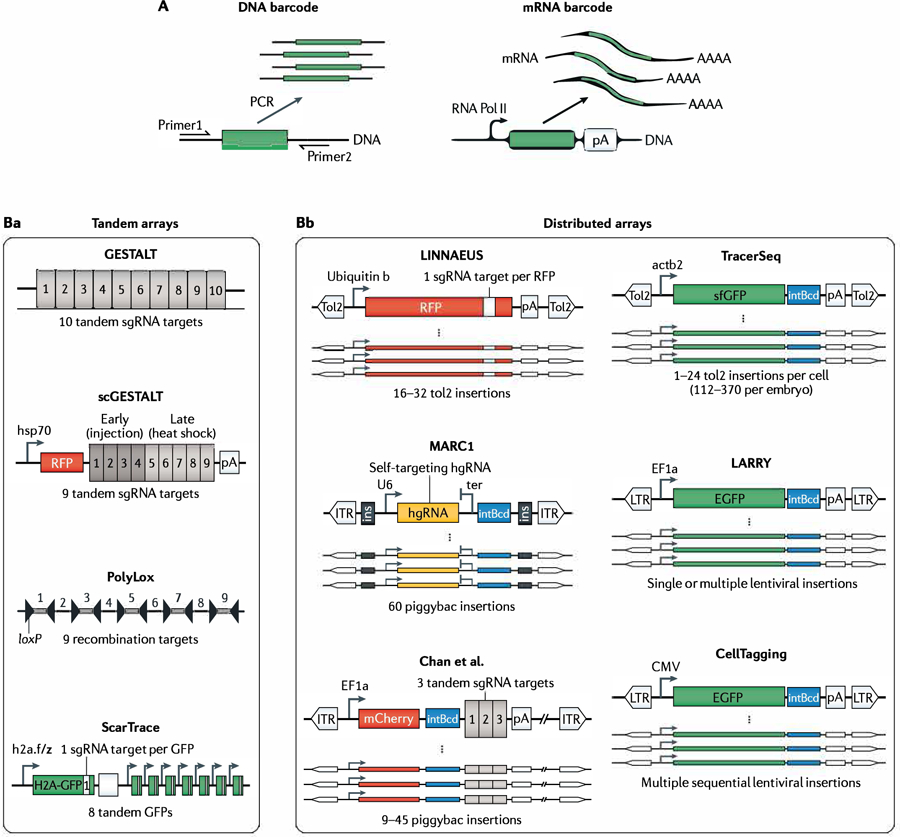
References
-
- Whitman CO Memoirs: the embryology of clepsine. J. Cell Sci s2-18, 215–315 (1878).
-
- Waddington CH The strategy of the genes A discussion of some aspects of theoretical biology. (George Allen & Unwin, Ltd., London, 1957).
-
- Saelens W, Cannoodt R, Todorov H & Saeys Y A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554 (2019). - PubMed
-
- Tritschler S et al. Concepts and limitations for learning developmental trajectories from single cell genomics. Development 146, dev170506 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
