

Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy
- PMID: 32238357
- PMCID: PMC7777391
- DOI: 10.1158/0008-5472.CAN-19-2959
Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy
Abstract
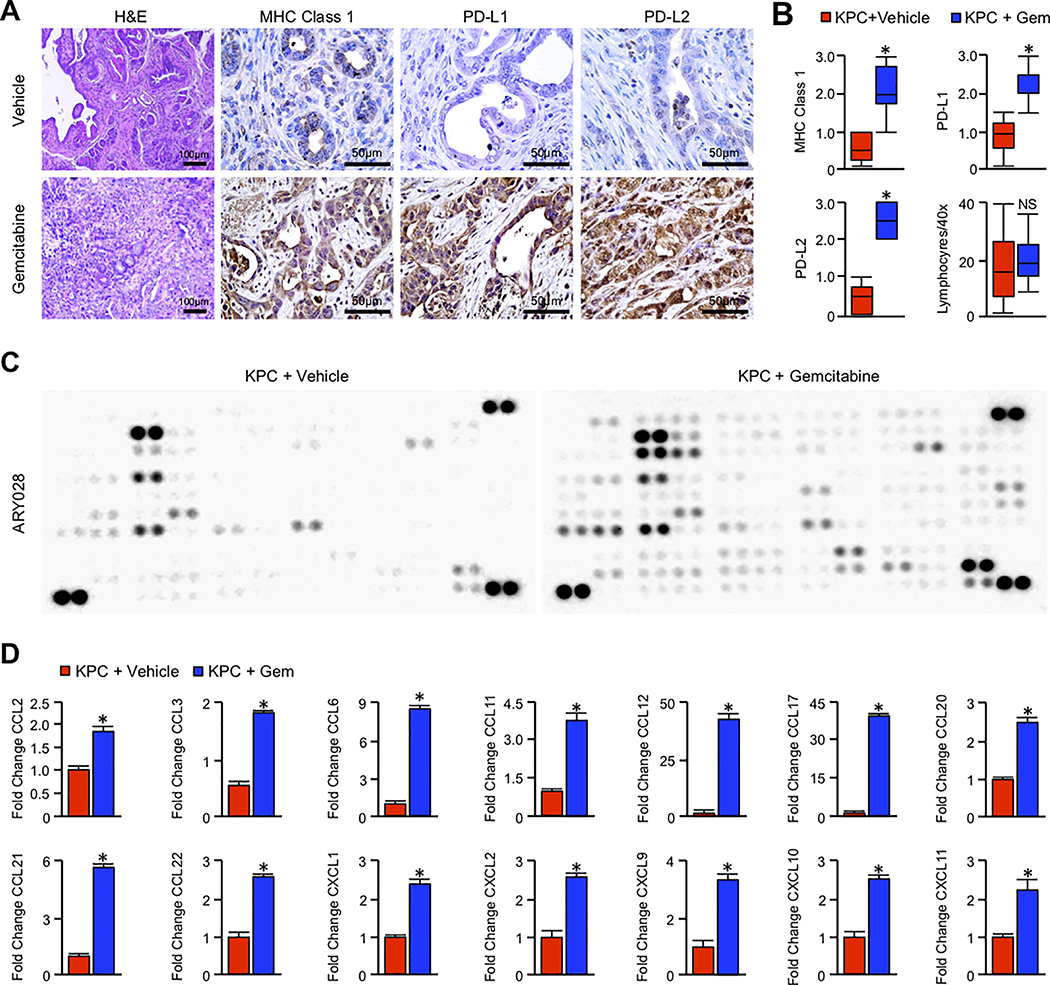
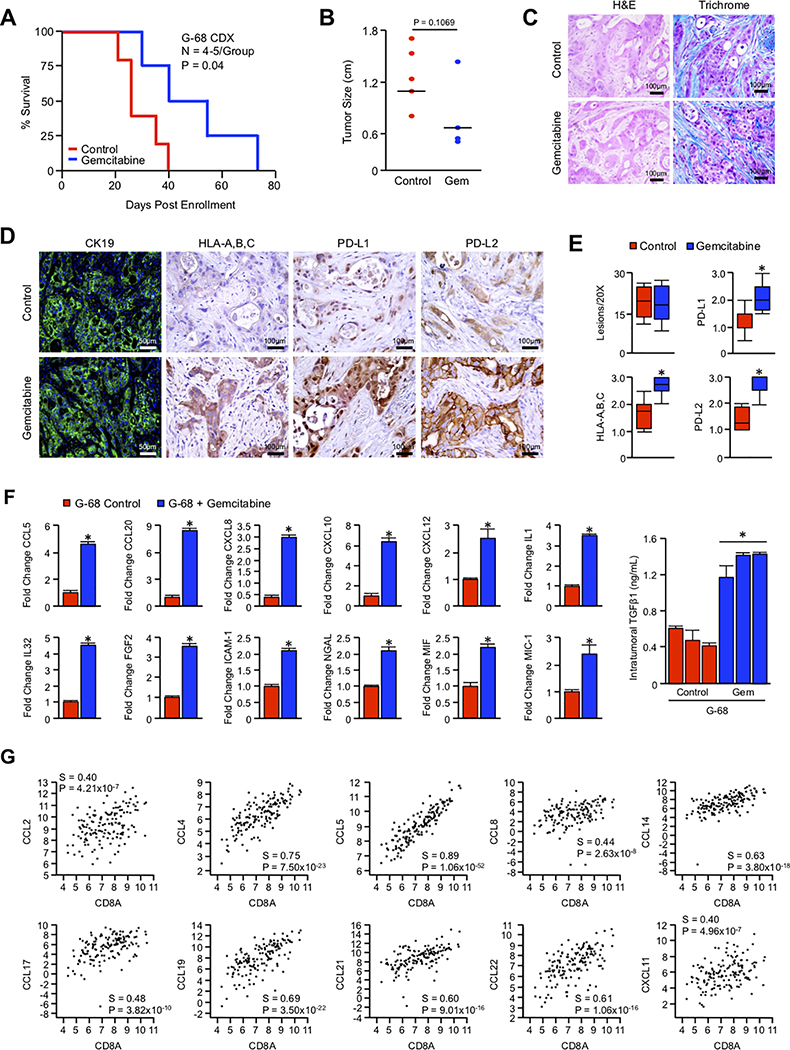
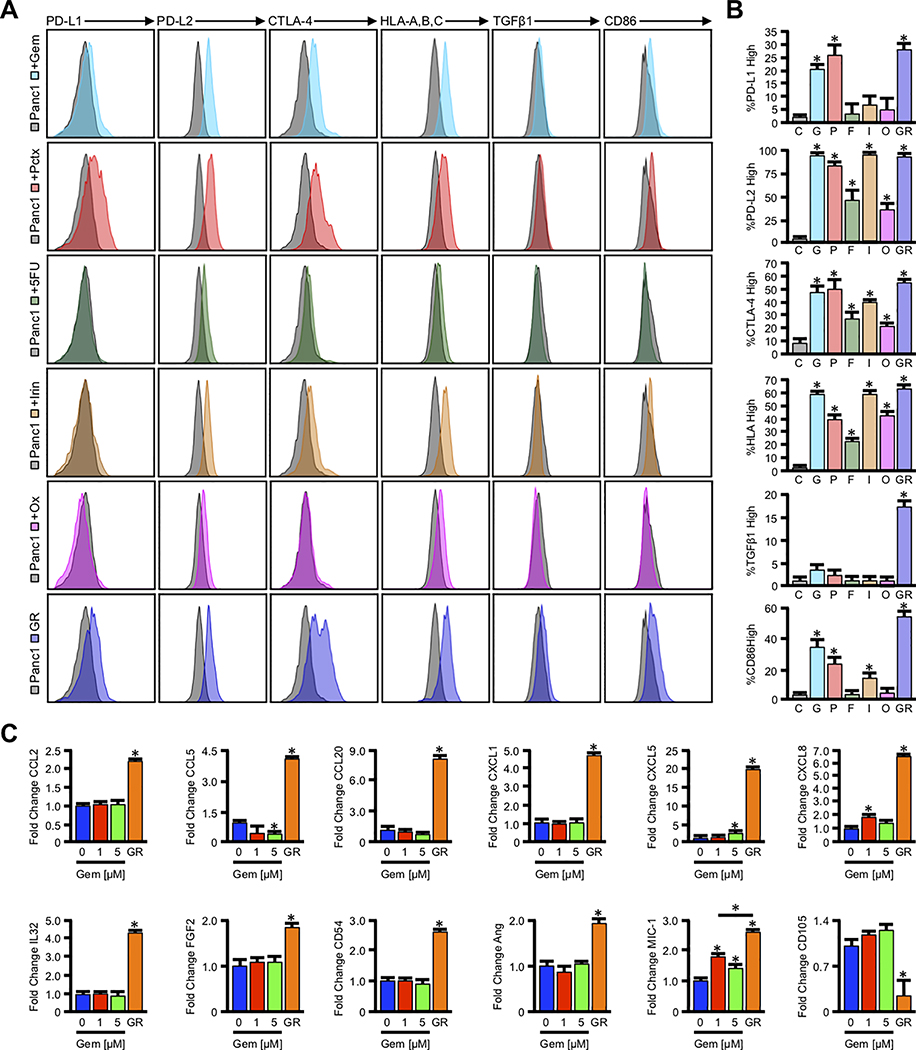
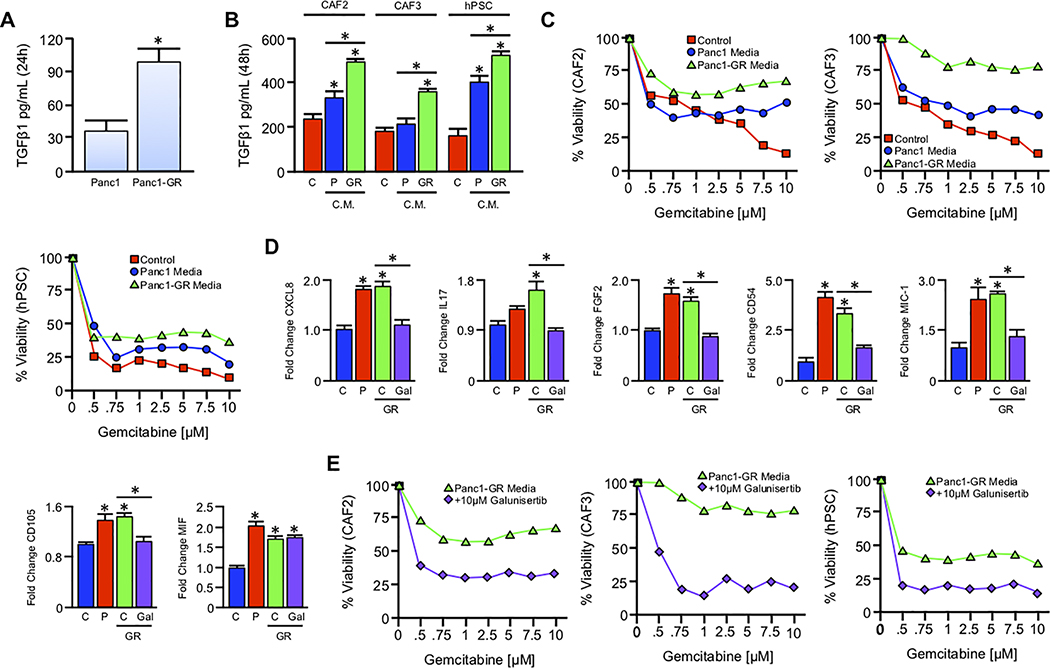
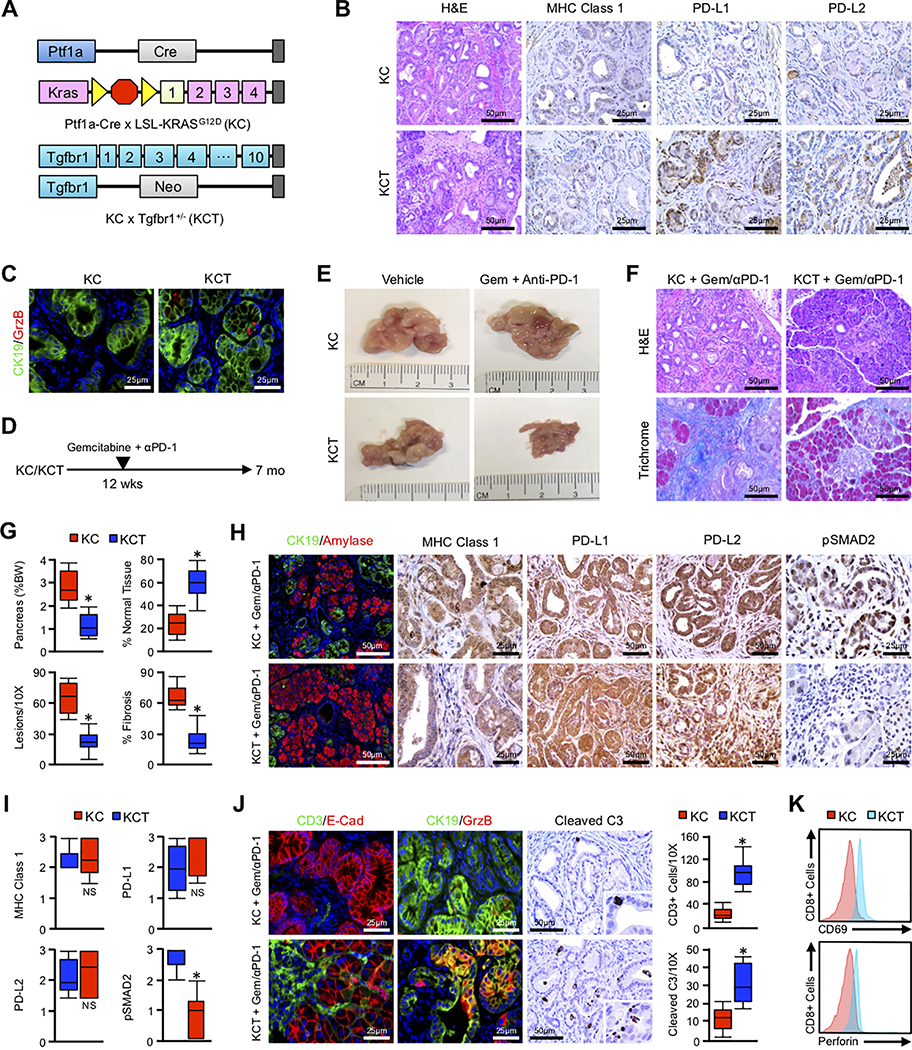
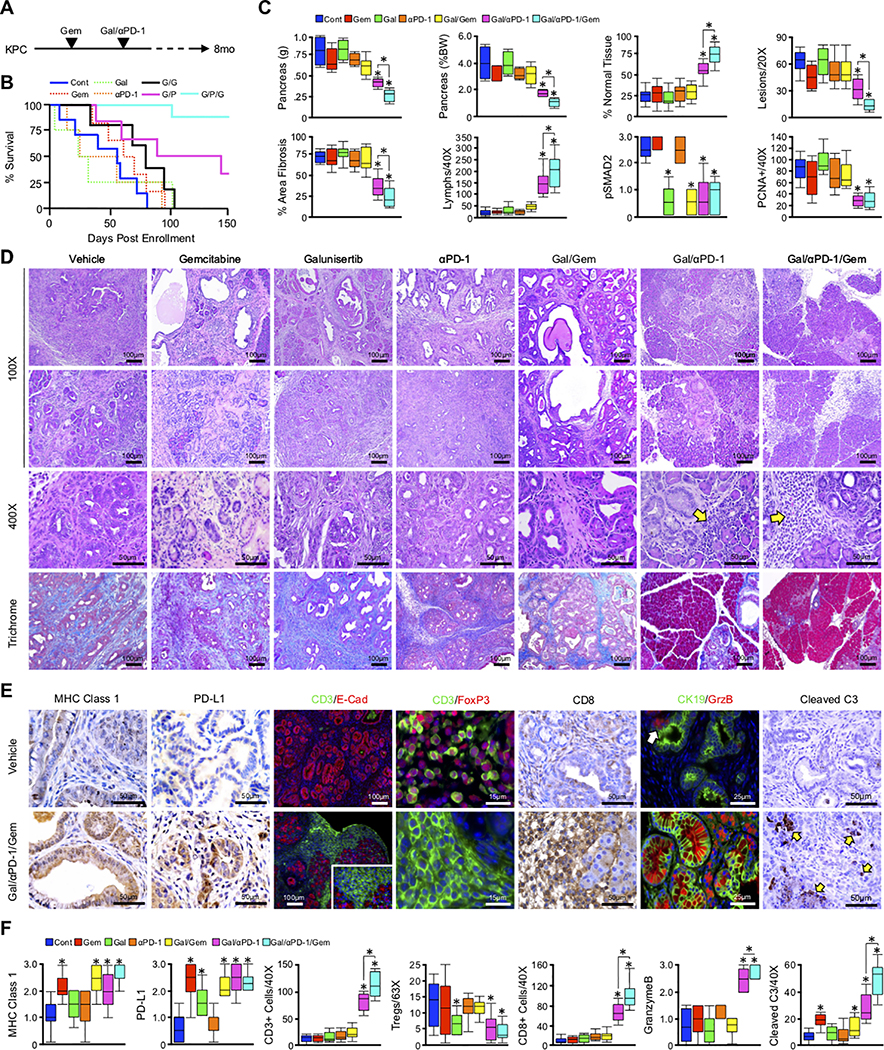
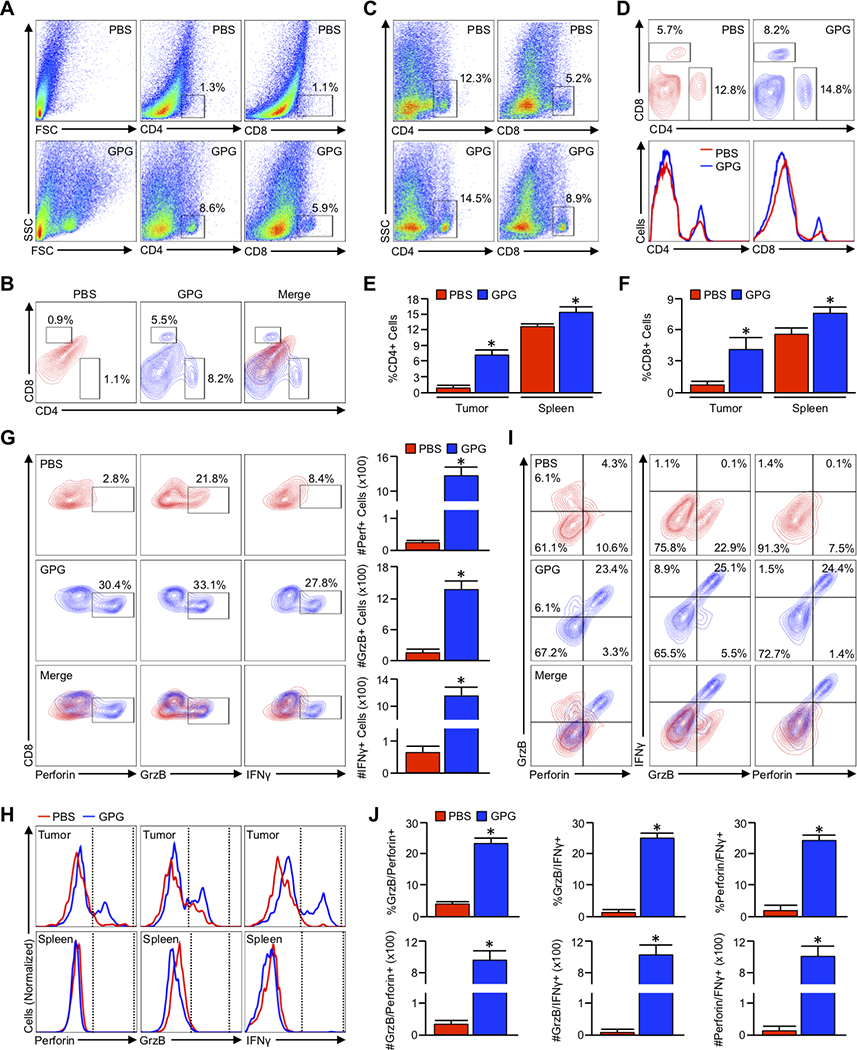
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer-related death with a median survival time of 6-12 months. Most patients present with disseminated disease and the majority are offered palliative chemotherapy. With no approved treatment modalities for patients who progress on chemotherapy, we explored the effects of long-term gemcitabine administration on the tumor microenvironment to identify potential therapeutic options for chemorefractory PDAC. Using a combination of mouse models, primary cell line-derived xenografts, and established tumor cell lines, we first evaluated chemotherapy-induced alterations in the tumor secretome and immune surface proteins by high throughput proteomic arrays. In addition to enhancing antigen presentation and immune checkpoint expression, gemcitabine consistently increased the synthesis of CCL/CXCL chemokines and TGFβ-associated signals. These secreted factors altered the composition of the tumor stroma, conferring gemcitabine resistance to cancer-associated fibroblasts in vitro and further enhancing TGFβ1 biosynthesis. Combined gemcitabine and anti-PD-1 treatment in transgenic models of murine PDAC failed to alter disease course unless mice also underwent genetic or pharmacologic ablation of TGFβ signaling. In the setting of TGFβ signaling deficiency, gemcitabine and anti-PD-1 led to a robust CD8+ T-cell response and decrease in tumor burden, markedly enhancing overall survival. These results suggest that gemcitabine successfully primes PDAC tumors for immune checkpoint inhibition by enhancing antigen presentation only following disruption of the immunosuppressive cytokine barrier. Given the current lack of third-line treatment options, this approach warrants consideration in the clinical management of gemcitabine-refractory PDAC. SIGNIFICANCE: These data suggest that long-term treatment with gemcitabine leads to extensive reprogramming of the pancreatic tumor microenvironment and that patients who progress on gemcitabine-based regimens may benefit from multidrug immunotherapy.See related commentary by Carpenter et al., p. 3070 GRAPHICAL ABSTRACT: http://cancerres.aacrjournals.org/content/canres/80/15/3101/F1.large.jpg.
©2020 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment in
-
Targeting the Microenvironment to Overcome Gemcitabine Resistance in Pancreatic Cancer.Cancer Res. 2020 Aug 1;80(15):3070-3071. doi: 10.1158/0008-5472.CAN-20-1692. Cancer Res. 2020. PMID: 32753486
Comment on
-
Targeting the Microenvironment to Overcome Gemcitabine Resistance in Pancreatic Cancer.Cancer Res. 2020 Aug 1;80(15):3070-3071. doi: 10.1158/0008-5472.CAN-20-1692. Cancer Res. 2020. PMID: 32753486
References
-
- Wang Z, et al. (2011) Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol 8(1):27–33. - PubMed
-
- Moore M (1996) Activity of gemcitabine in patients with advanced pancreatic carcinoma. A review. Cancer 78(3 Suppl):633–638. - PubMed
-
- Casper ES, et al. (1994) Phase II trial of gemcitabine (2,2’-difluorodeoxycytidine) in patients with adenocarcinoma of the pancreas. Invest New Drugs 12(1):29–34. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
