

CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities
- PMID: 32238925
- PMCID: PMC7368463
- DOI: 10.1038/s41586-020-2099-x
CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities
Abstract
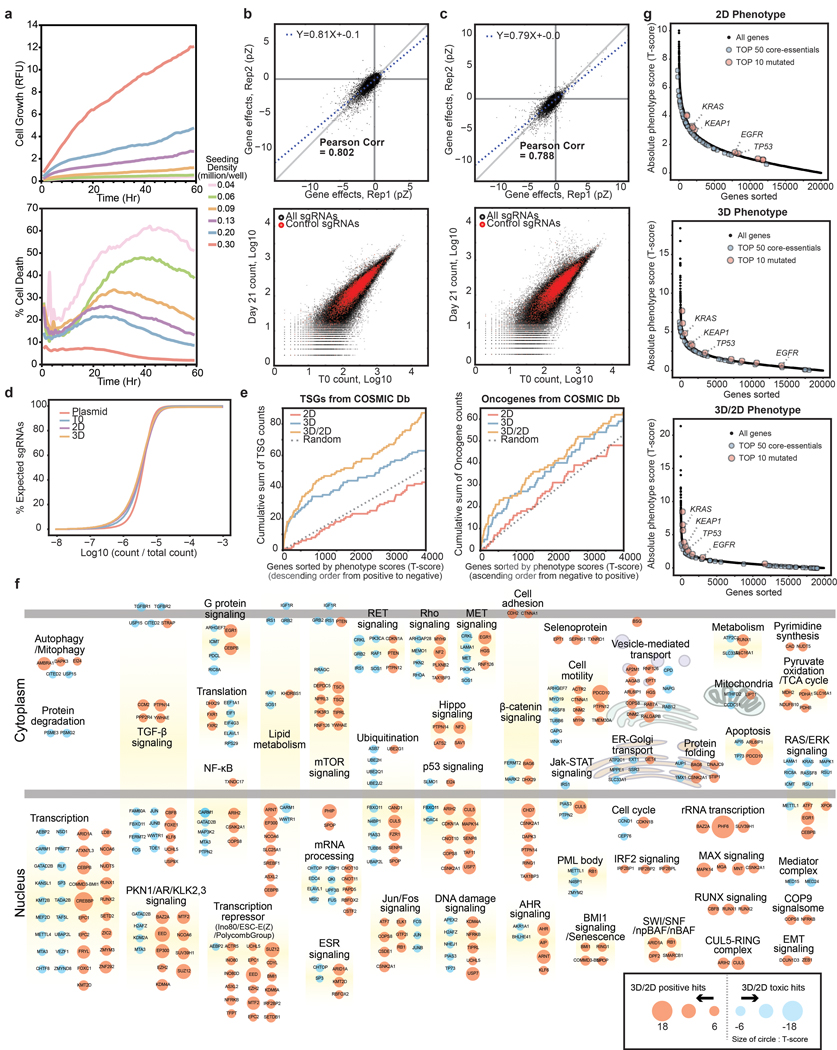
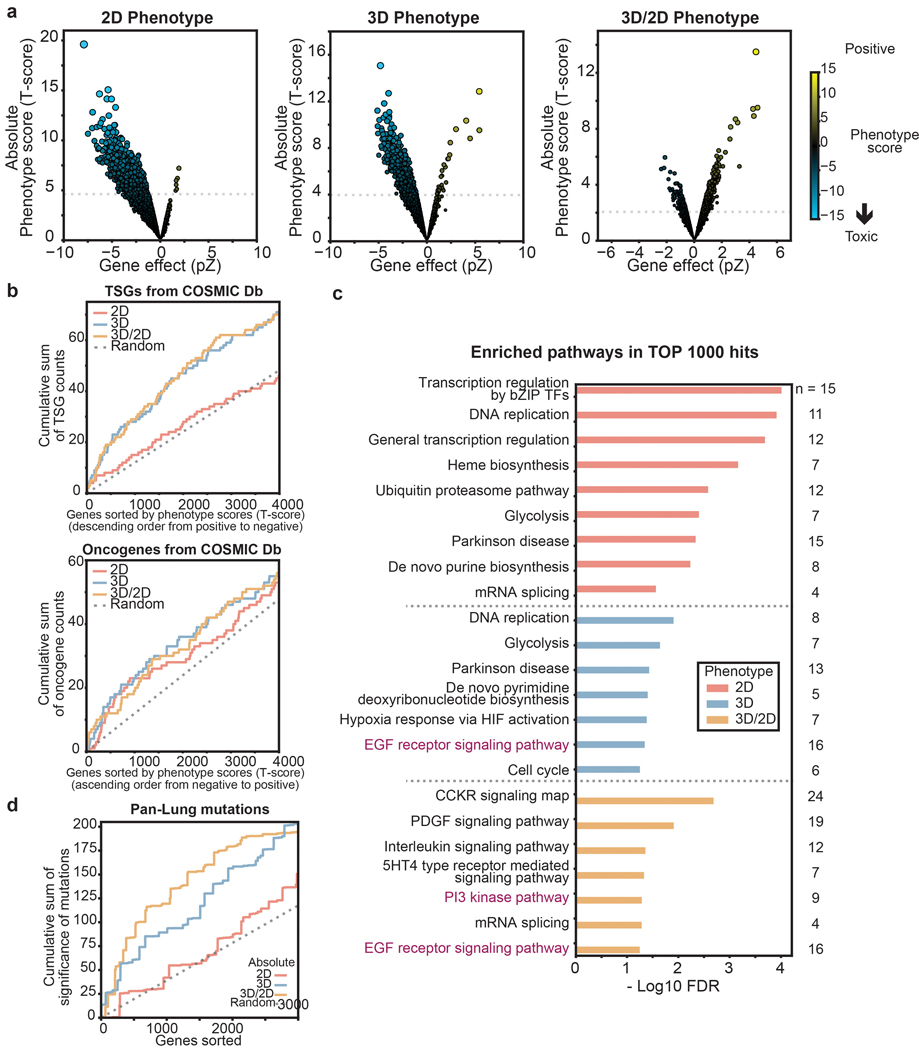
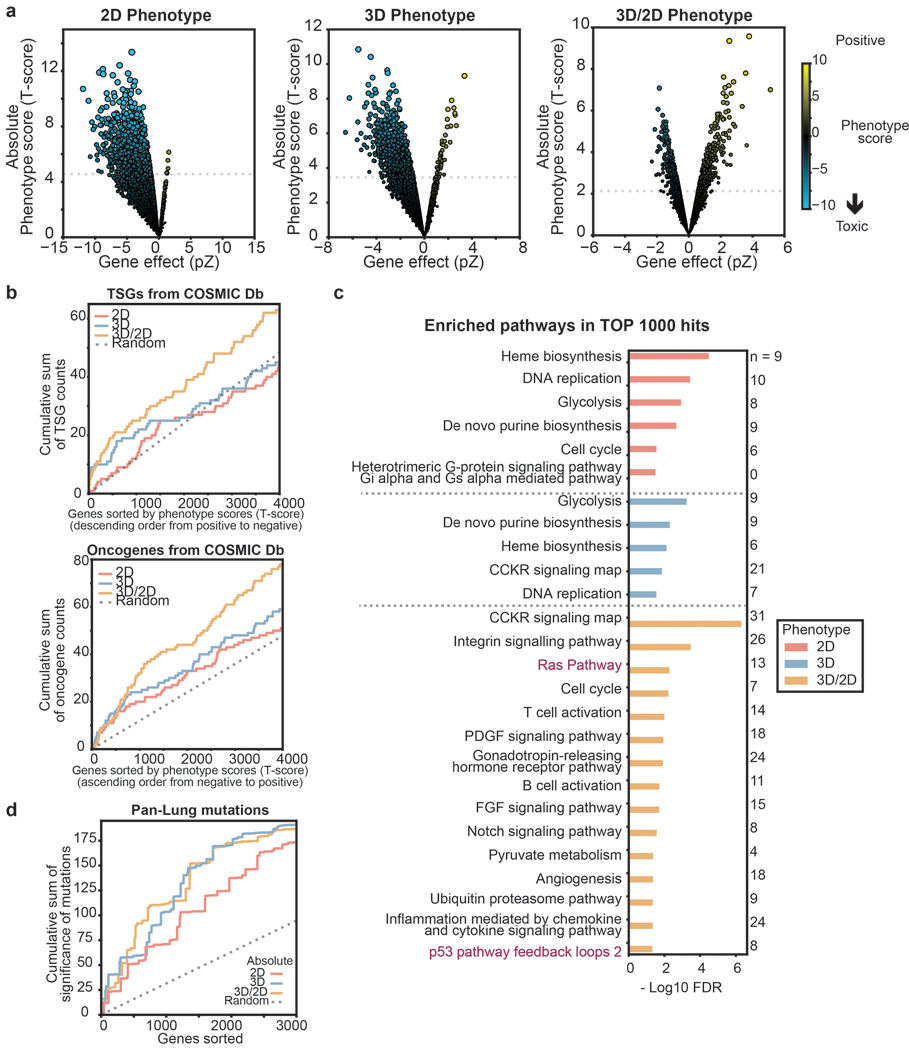
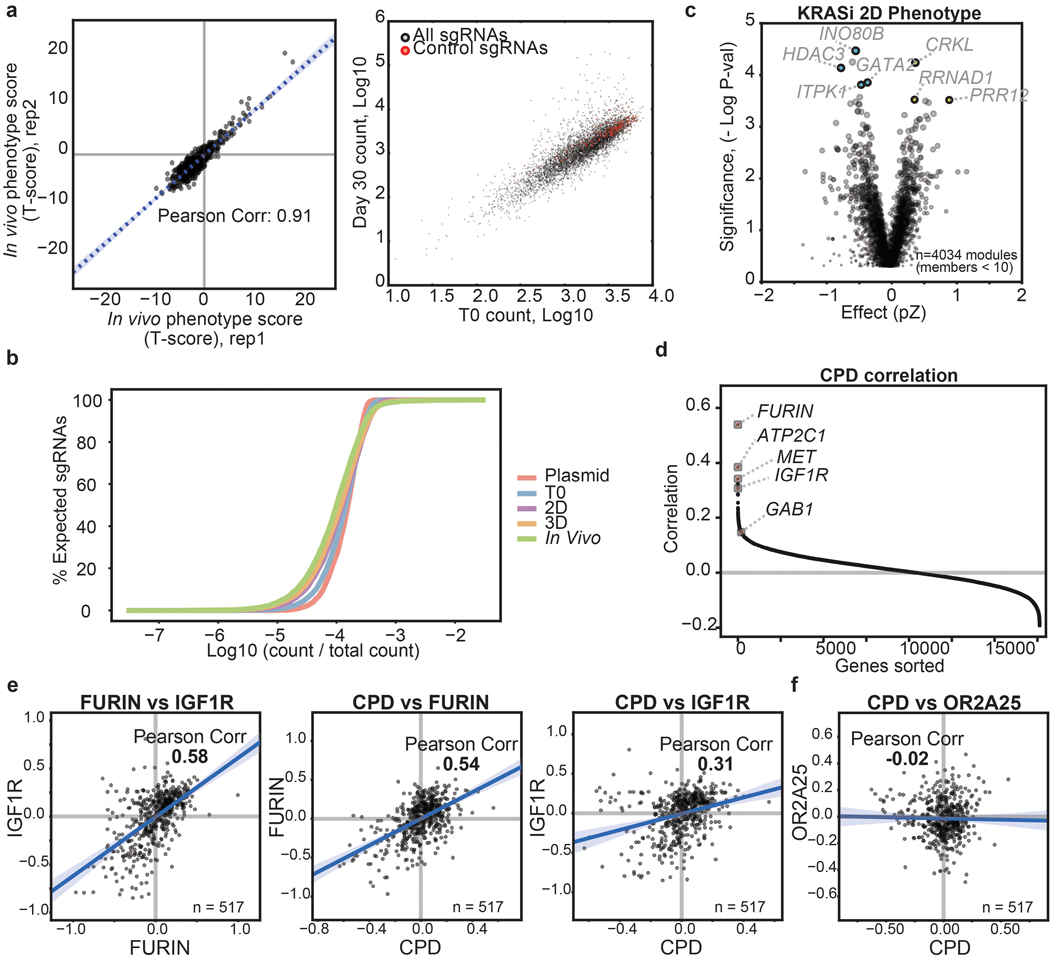
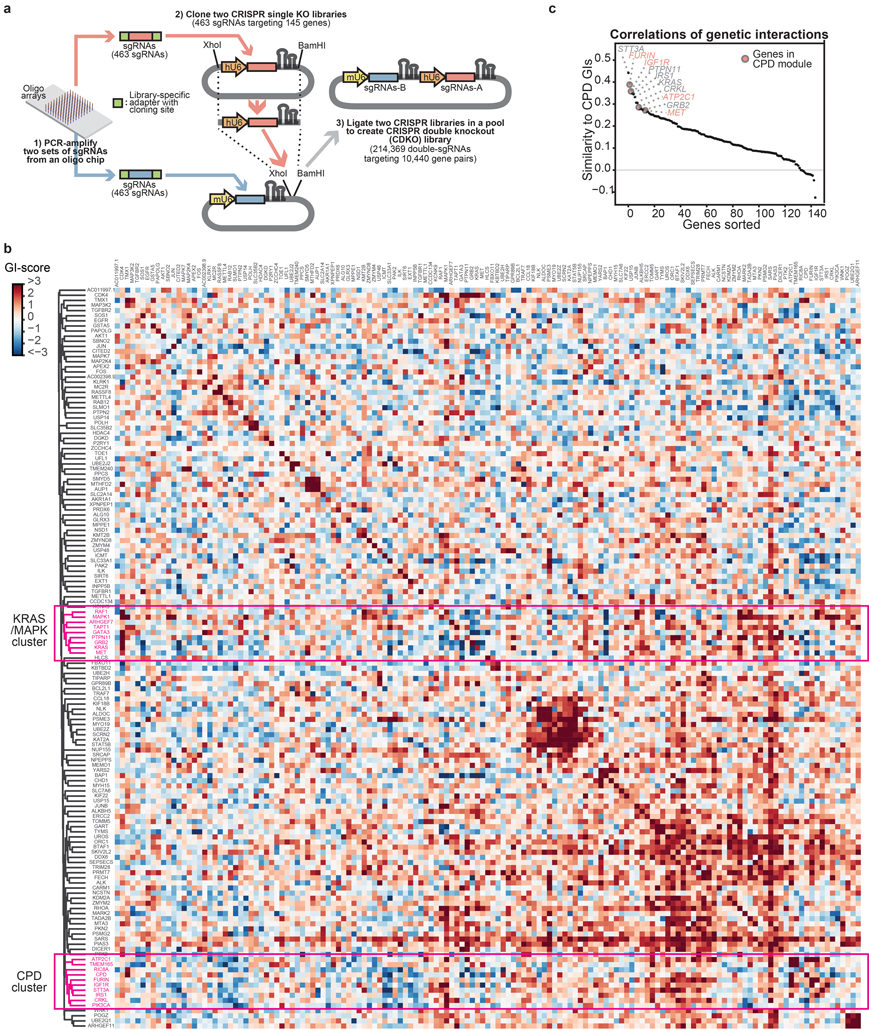
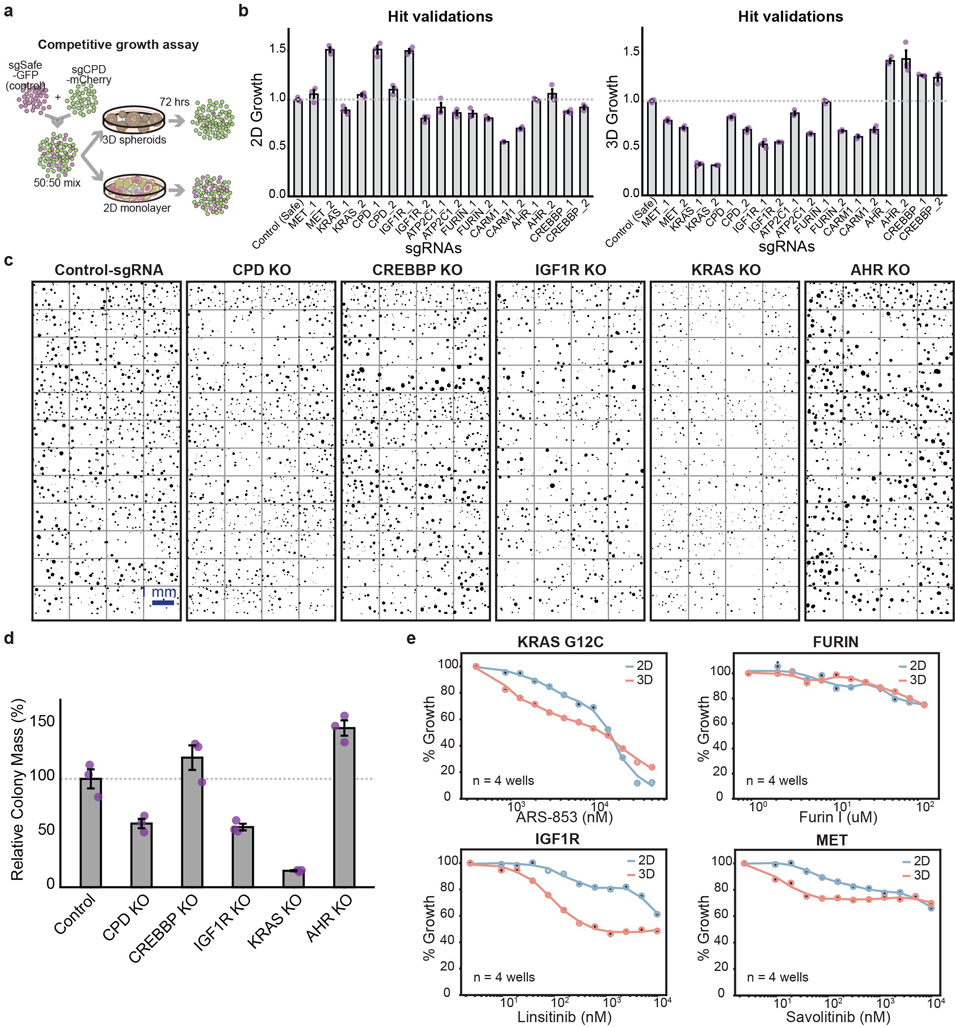
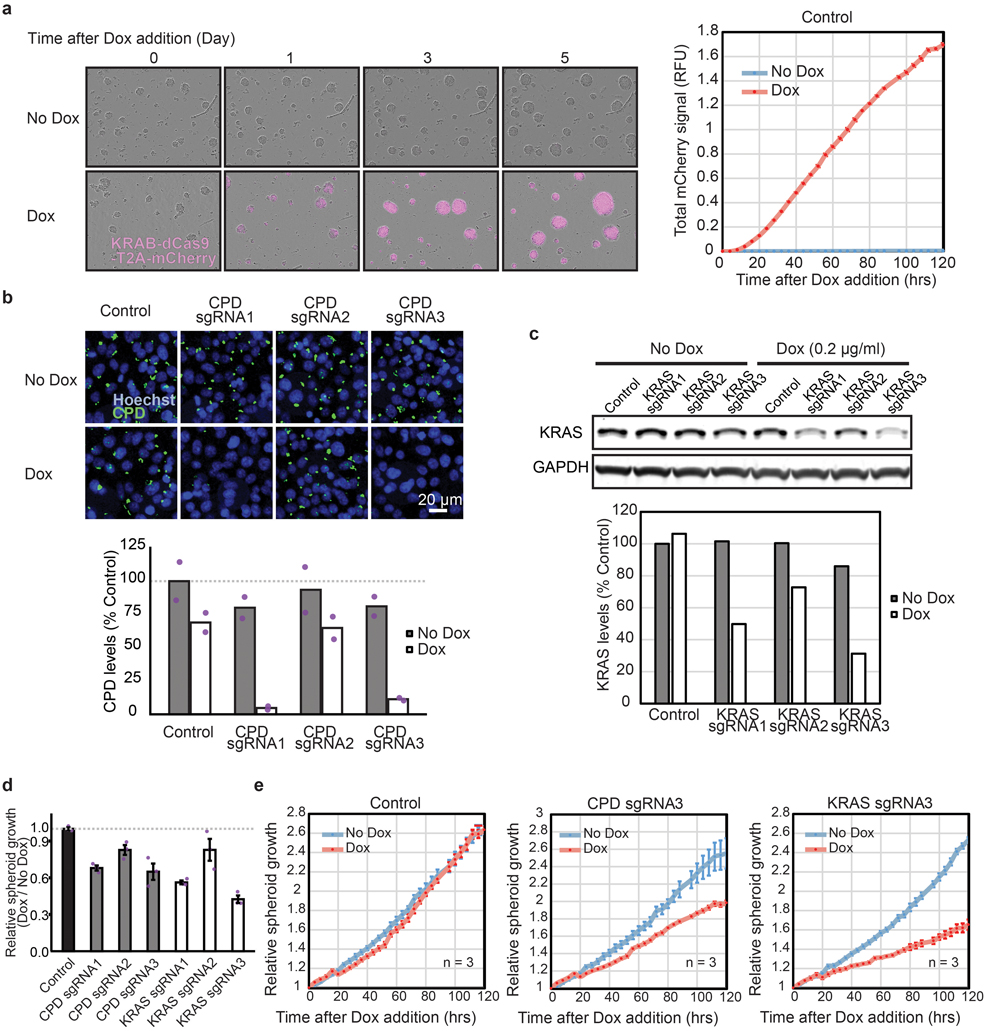
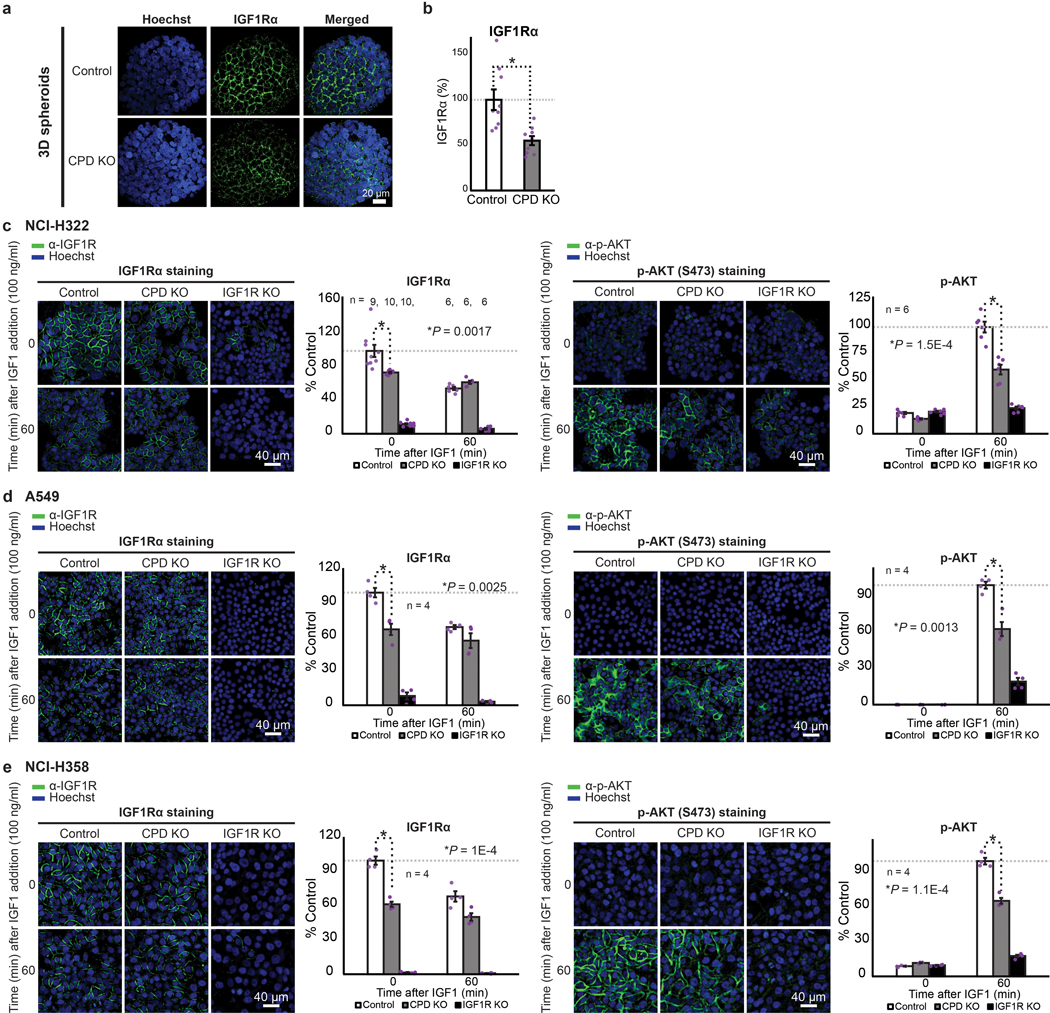
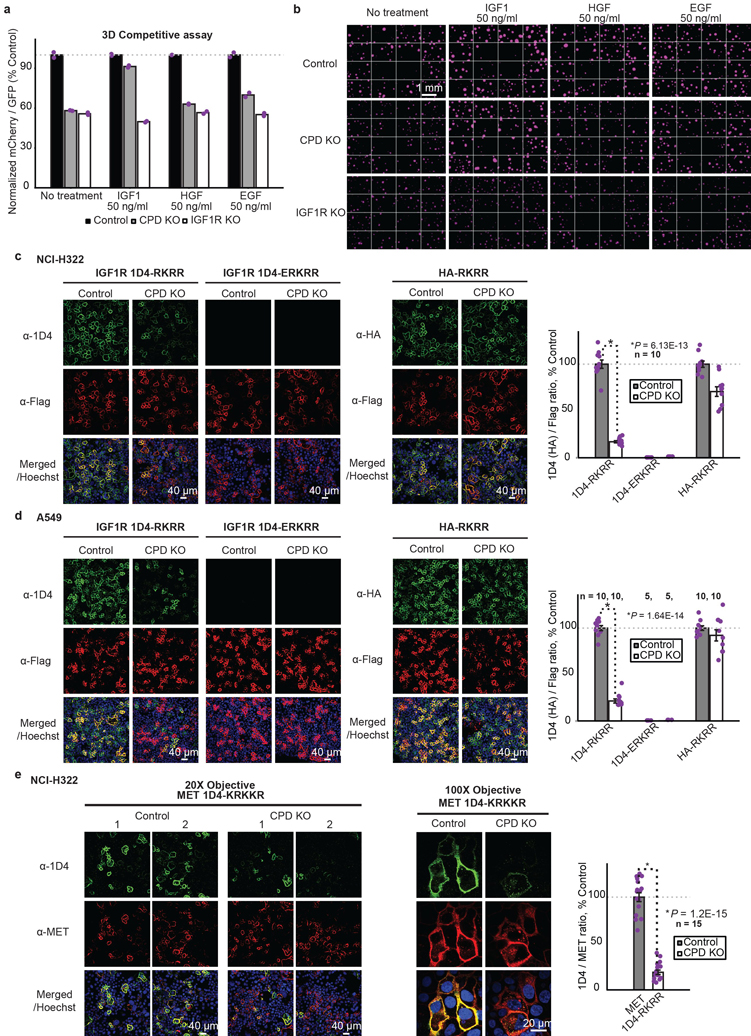
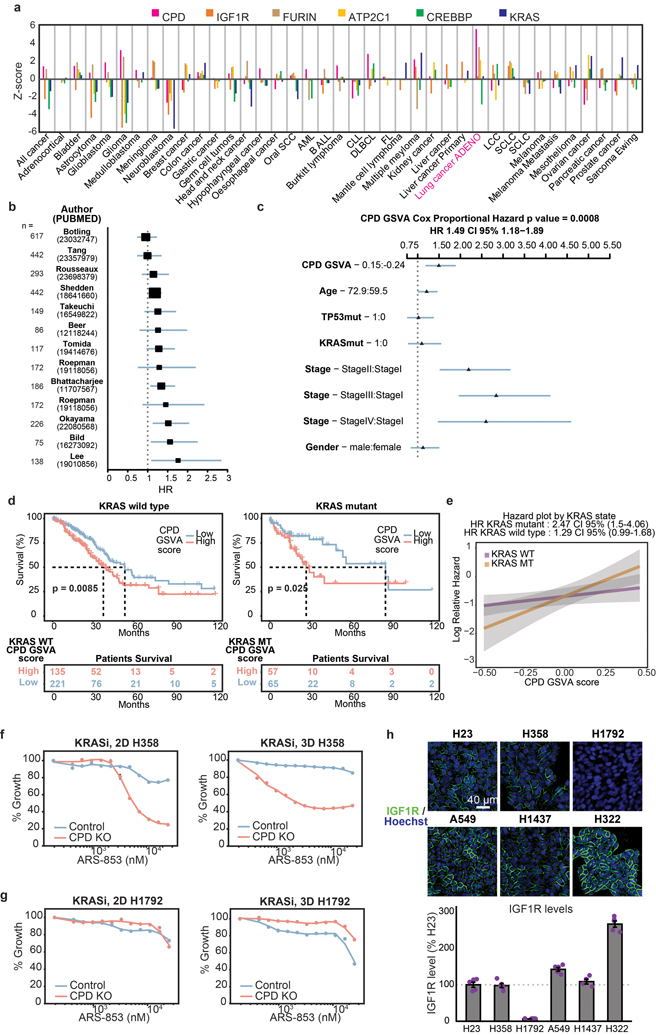
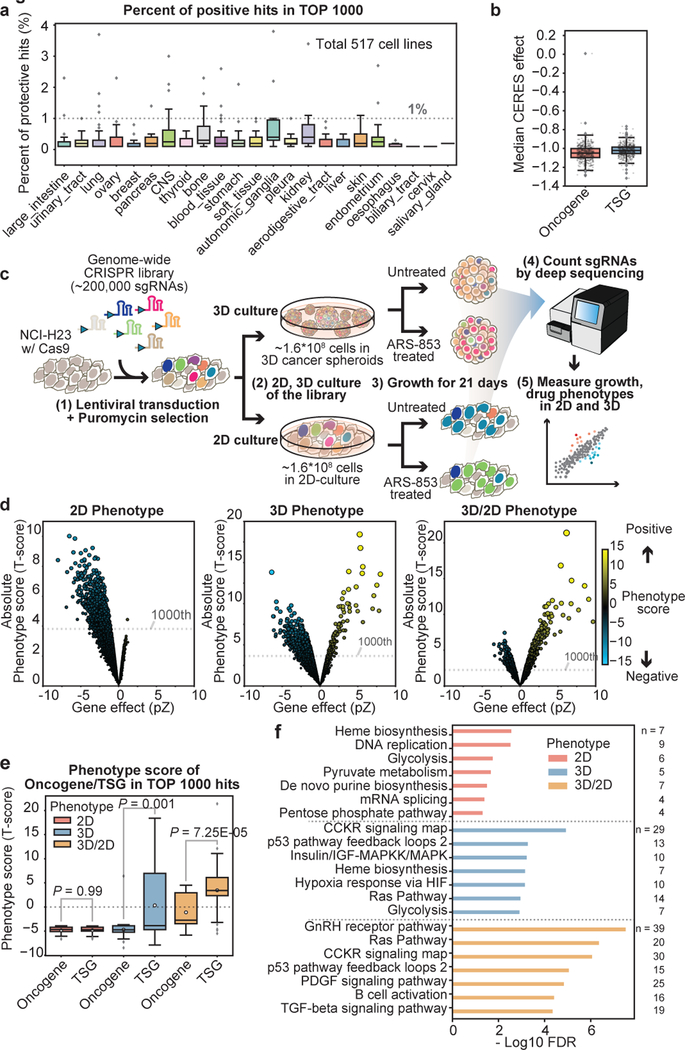
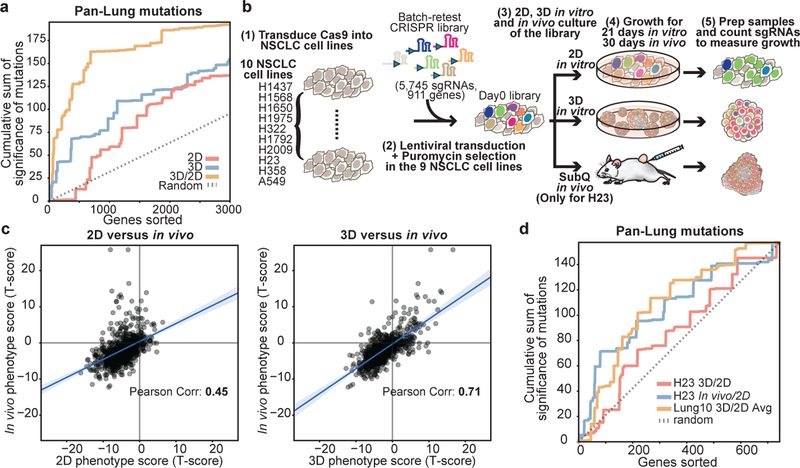
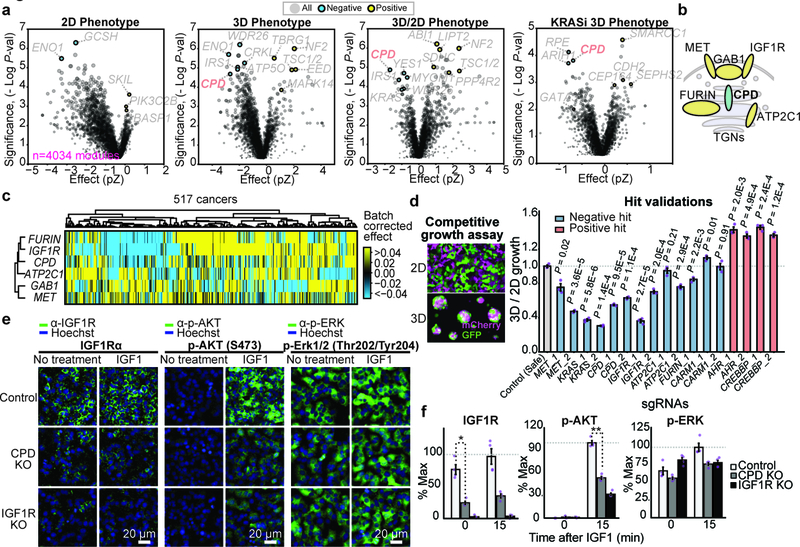
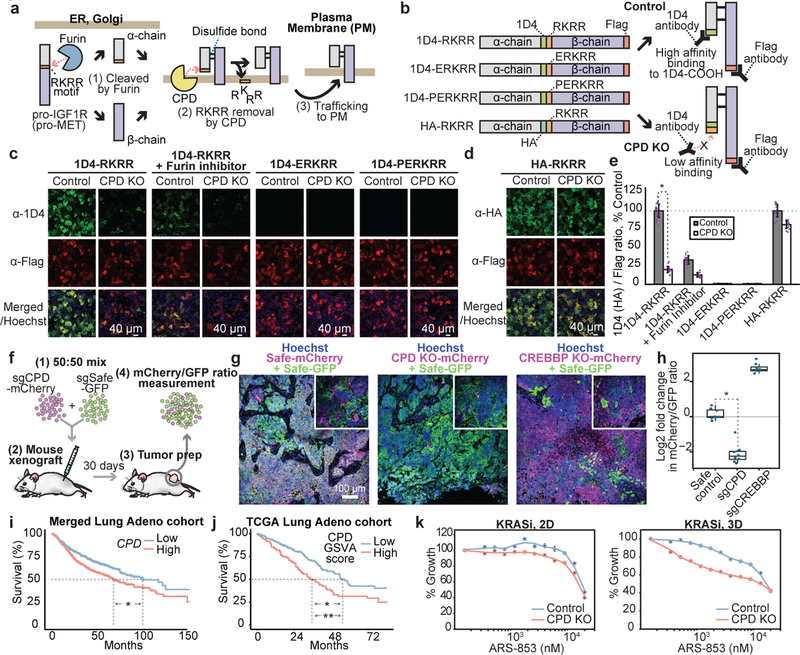
Cancer genomics studies have identified thousands of putative cancer driver genes1. Development of high-throughput and accurate models to define the functions of these genes is a major challenge. Here we devised a scalable cancer-spheroid model and performed genome-wide CRISPR screens in 2D monolayers and 3D lung-cancer spheroids. CRISPR phenotypes in 3D more accurately recapitulated those of in vivo tumours, and genes with differential sensitivities between 2D and 3D conditions were highly enriched for genes that are mutated in lung cancers. These analyses also revealed drivers that are essential for cancer growth in 3D and in vivo, but not in 2D. Notably, we found that carboxypeptidase D is responsible for removal of a C-terminal RKRR motif2 from the α-chain of the insulin-like growth factor 1 receptor that is critical for receptor activity. Carboxypeptidase D expression correlates with patient outcomes in patients with lung cancer, and loss of carboxypeptidase D reduced tumour growth. Our results reveal key differences between 2D and 3D cancer models, and establish a generalizable strategy for performing CRISPR screens in spheroids to reveal cancer vulnerabilities.
Conflict of interest statement
ETHICS DECLARATIONS
Competing interests
Authors through the Office of Technology Licensing at Stanford University have filed patent applications on methods for inhibiting tumor growth by inhibiting CPD as well as systems and methods for identifying CPD inhibitors and other tumor suppressors and/or oncogenes.
Figures
References
-
- Garraway LA & Lander ES Lessons from the cancer genome. Cell 153, 17–37 (2013). - PubMed
-
- Khatib A-M et al. Inhibition of proprotein convertases is associated with loss of growth and tumorigenicity of HT-29 human colon carcinoma cells importance of insulin-like growth factor-1 (IGF-1) receptor processing in IGF-1-mediated functions. J. Biol. Chem 276, 30686–30693 (2001). - PubMed
-
- Unger C et al. Modeling human carcinomas: physiologically relevant 3D models to improve anti-cancer drug development. Adv. Drug Deliv. Rev 79–80, 50–67 (2014). - PubMed
-
- Sharpless NE & Depinho RA The mighty mouse: genetically engineered mouse models in cancer drug development. Nat. Rev. Drug Discov. 5, 741–754 (2006). - PubMed
REFERENCES FOR METHODS
-
- Bray NL, Pimentel H, Melsted P & Pachter L. Erratum: Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 888 (2016). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
