

Two Synthetic 18-Way Outcrossed Populations of Diploid Budding Yeast with Utility for Complex Trait Dissection
- PMID: 32241804
- PMCID: PMC7268983
- DOI: 10.1534/genetics.120.303202
Two Synthetic 18-Way Outcrossed Populations of Diploid Budding Yeast with Utility for Complex Trait Dissection
Abstract
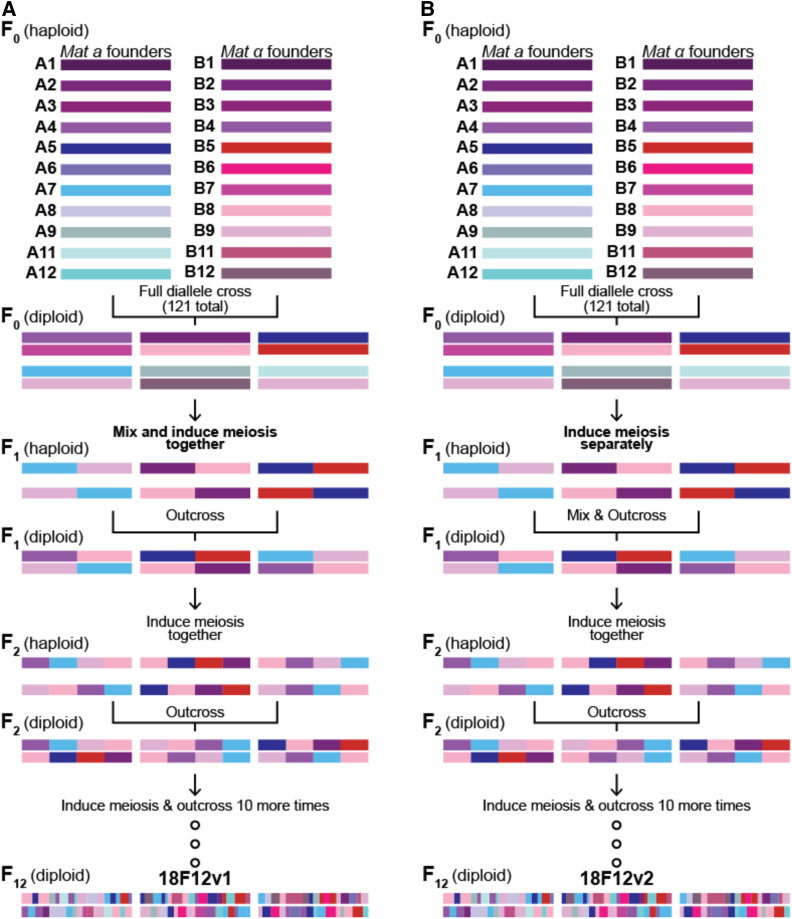
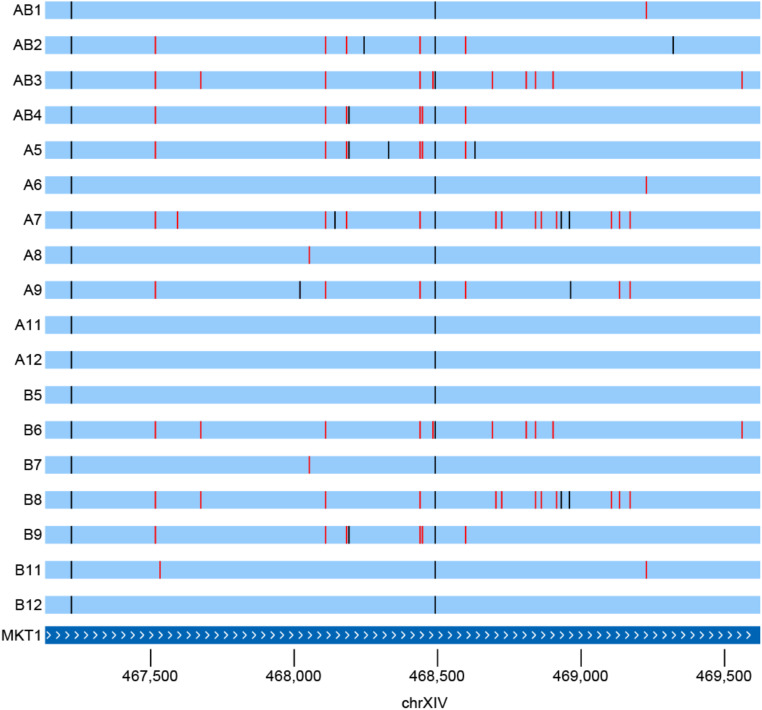
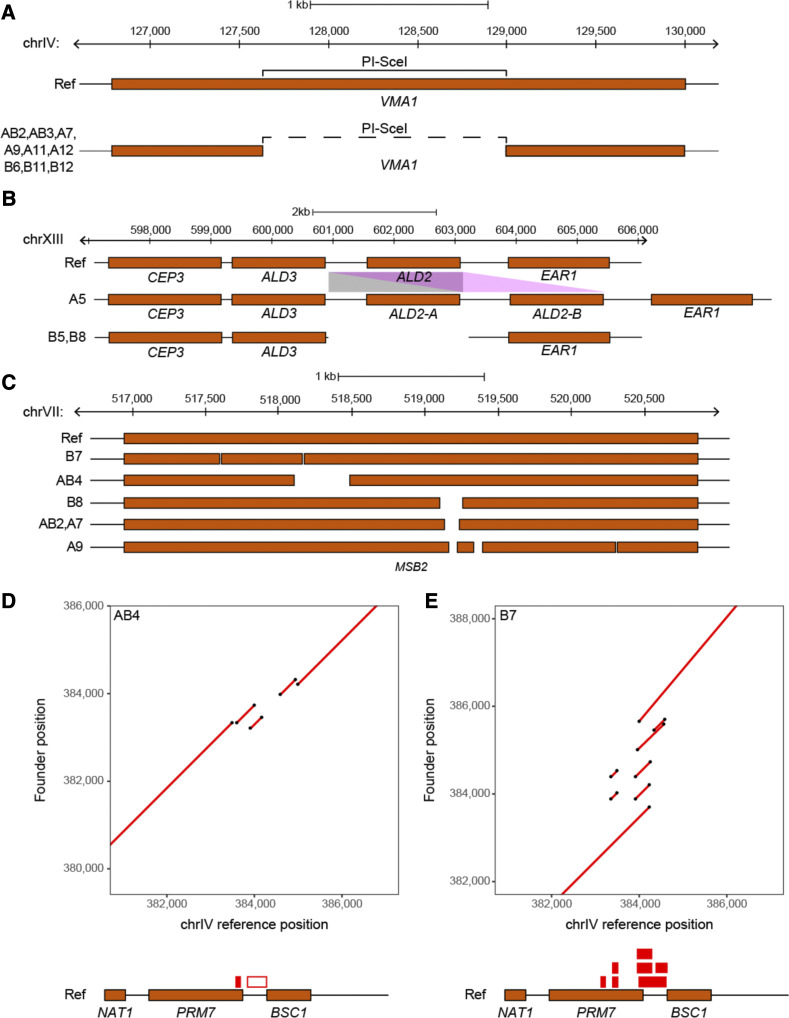
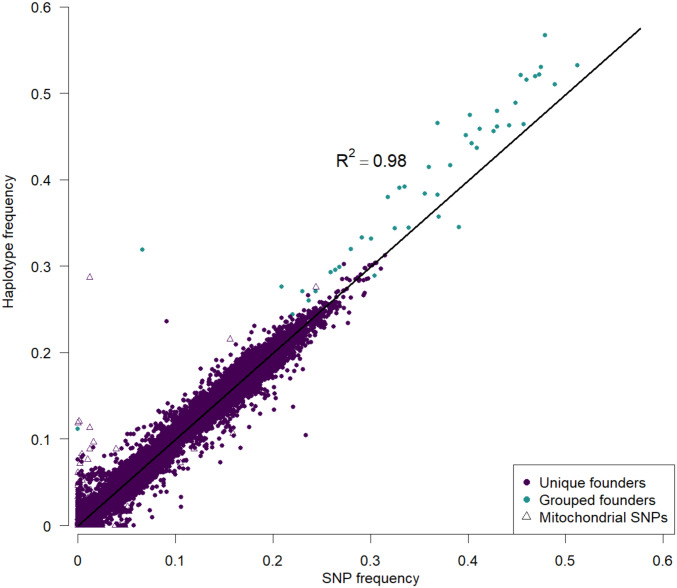
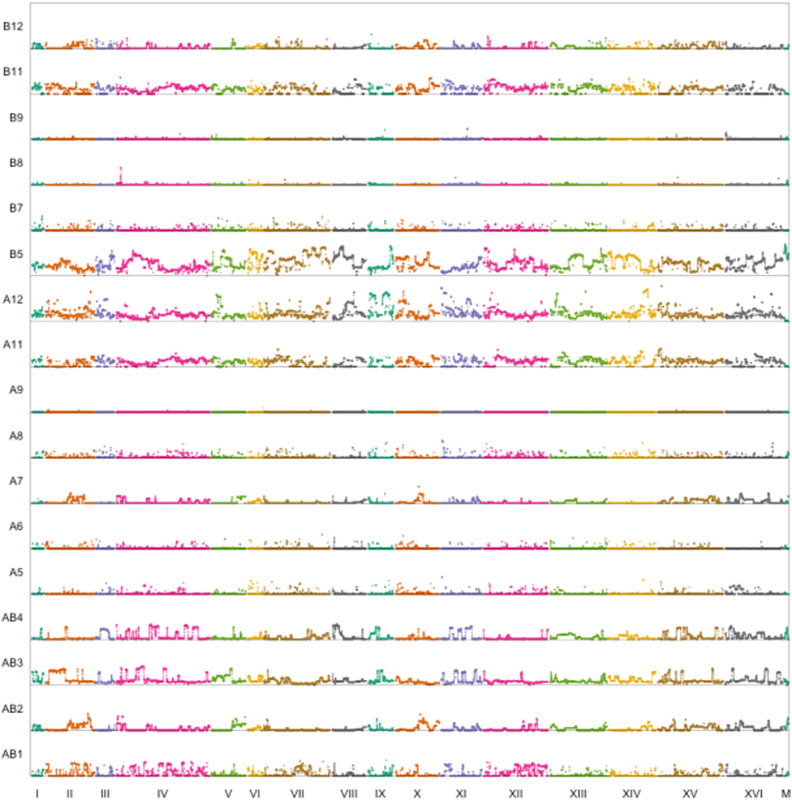
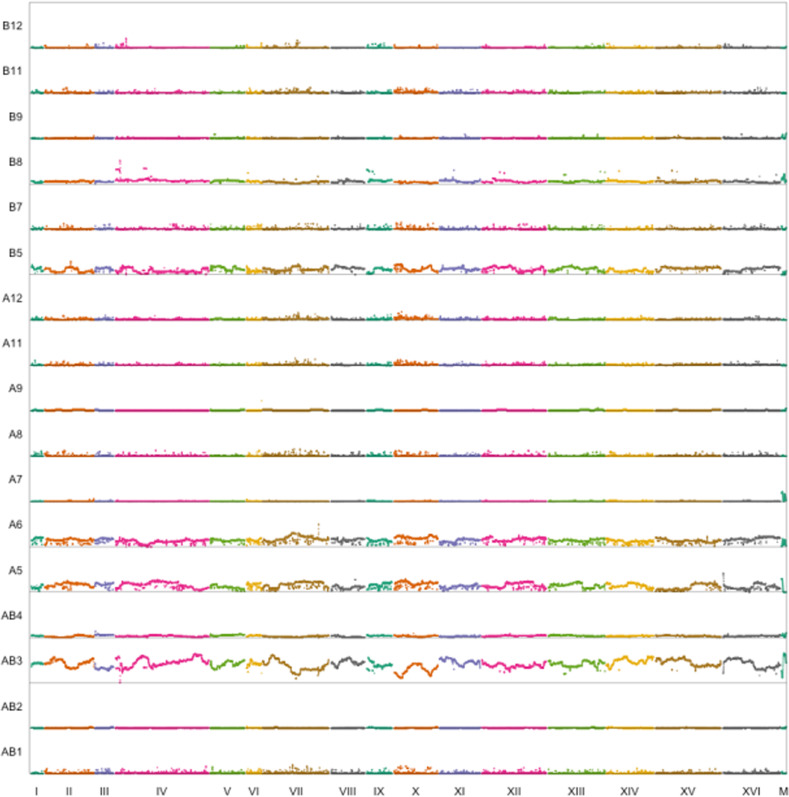
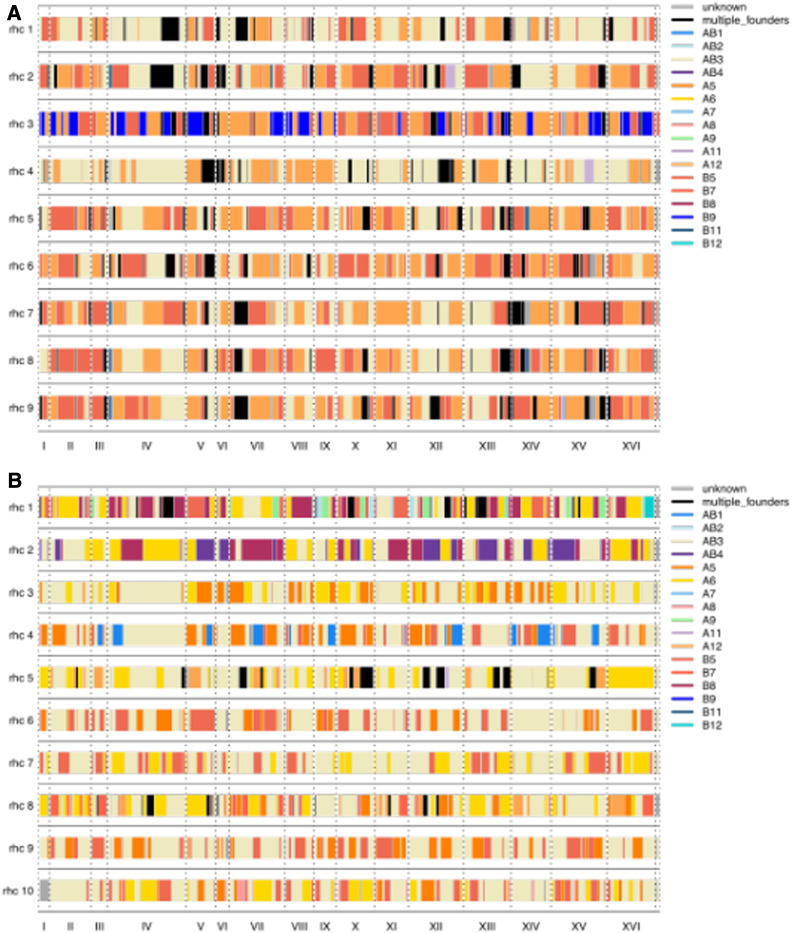
Advanced-generation multiparent populations (MPPs) are a valuable tool for dissecting complex traits, having more power than genome-wide association studies to detect rare variants and higher resolution than F2 linkage mapping. To extend the advantages of MPPs in budding yeast, we describe the creation and characterization of two outbred MPPs derived from 18 genetically diverse founding strains. We carried out de novo assemblies of the genomes of the 18 founder strains, such that virtually all variation segregating between these strains is known, and represented those assemblies as Santa Cruz Genome Browser tracks. We discovered complex patterns of structural variation segregating among the founders, including a large deletion within the vacuolar ATPase VMA1, several different deletions within the osmosensor MSB2, a series of deletions and insertions at PRM7 and the adjacent BSC1, as well as copy number variation at the dehydrogenase ALD2 Resequenced haploid recombinant clones from the two MPPs have a median unrecombined block size of 66 kb, demonstrating that the population is highly recombined. We pool-sequenced the two MPPs to 3270× and 2226× coverage and demonstrated that we can accurately estimate local haplotype frequencies using pooled data. We further downsampled the pool-sequenced data to ∼20-40× and showed that local haplotype frequency estimates remained accurate, with median error rates 0.8 and 0.6% at 20× and 40×, respectively. Haplotypes frequencies are estimated much more accurately than SNP frequencies obtained directly from the same data. Deep sequencing of the two populations revealed that 10 or more founders are present at a detectable frequency for > 98% of the genome, validating the utility of this resource for the exploration of the role of standing variation in the architecture of complex traits.
Keywords: MPP; Multiparent Advanced Generation Inter-Cross (MAGIC); budding yeast; de novo assembly; haplotype inference; multiparental populations.
Copyright © 2020 by the Genetics Society of America.
Figures
Similar articles
-
An eight-parent multiparent advanced generation inter-cross population for winter-sown wheat: creation, properties, and validation.G3 (Bethesda). 2014 Sep 18;4(9):1603-10. doi: 10.1534/g3.114.012963. G3 (Bethesda). 2014. PMID: 25237112 Free PMC article.
-
Genomes of the Mouse Collaborative Cross.Genetics. 2017 Jun;206(2):537-556. doi: 10.1534/genetics.116.198838. Genetics. 2017. PMID: 28592495 Free PMC article.
-
Modeling allelic diversity of multiparent mapping populations affects detection of quantitative trait loci.G3 (Bethesda). 2022 Mar 4;12(3):jkac011. doi: 10.1093/g3journal/jkac011. G3 (Bethesda). 2022. PMID: 35100382 Free PMC article.
-
Facilitating Complex Trait Analysis via Reduced Complexity Crosses.Trends Genet. 2020 Aug;36(8):549-562. doi: 10.1016/j.tig.2020.05.003. Epub 2020 May 29. Trends Genet. 2020. PMID: 32482413 Free PMC article. Review.
-
Multiparent intercross populations in analysis of quantitative traits.J Genet. 2012;91(1):111-7. doi: 10.1007/s12041-012-0144-8. J Genet. 2012. PMID: 22546834 Review.
Cited by
-
A widespread inversion polymorphism conserved among Saccharomyces species is caused by recurrent homogenization of a sporulation gene family.PLoS Genet. 2022 Nov 28;18(11):e1010525. doi: 10.1371/journal.pgen.1010525. eCollection 2022 Nov. PLoS Genet. 2022. PMID: 36441813 Free PMC article.
-
ScRAPdb: an integrated pan-omics database for the Saccharomyces cerevisiae reference assembly panel.Nucleic Acids Res. 2025 Jan 6;53(D1):D852-D863. doi: 10.1093/nar/gkae955. Nucleic Acids Res. 2025. PMID: 39470715 Free PMC article.
-
Genetic Background Matters: Population-Based Studies in Model Organisms for Translational Research.Int J Mol Sci. 2022 Jul 8;23(14):7570. doi: 10.3390/ijms23147570. Int J Mol Sci. 2022. PMID: 35886916 Free PMC article. Review.
-
Discovery of malathion resistance QTL in Drosophila melanogaster using a bulked phenotyping approach.G3 (Bethesda). 2022 Dec 1;12(12):jkac279. doi: 10.1093/g3journal/jkac279. G3 (Bethesda). 2022. PMID: 36250804 Free PMC article.
-
Crossing design shapes patterns of genetic variation in synthetic recombinant populations of Saccharomyces cerevisiae.Sci Rep. 2021 Oct 1;11(1):19551. doi: 10.1038/s41598-021-99026-0. Sci Rep. 2021. PMID: 34599243 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
