

Structural basis for allosteric PARP-1 retention on DNA breaks
- PMID: 32241924
- PMCID: PMC7347020
- DOI: 10.1126/science.aax6367
Structural basis for allosteric PARP-1 retention on DNA breaks
Abstract
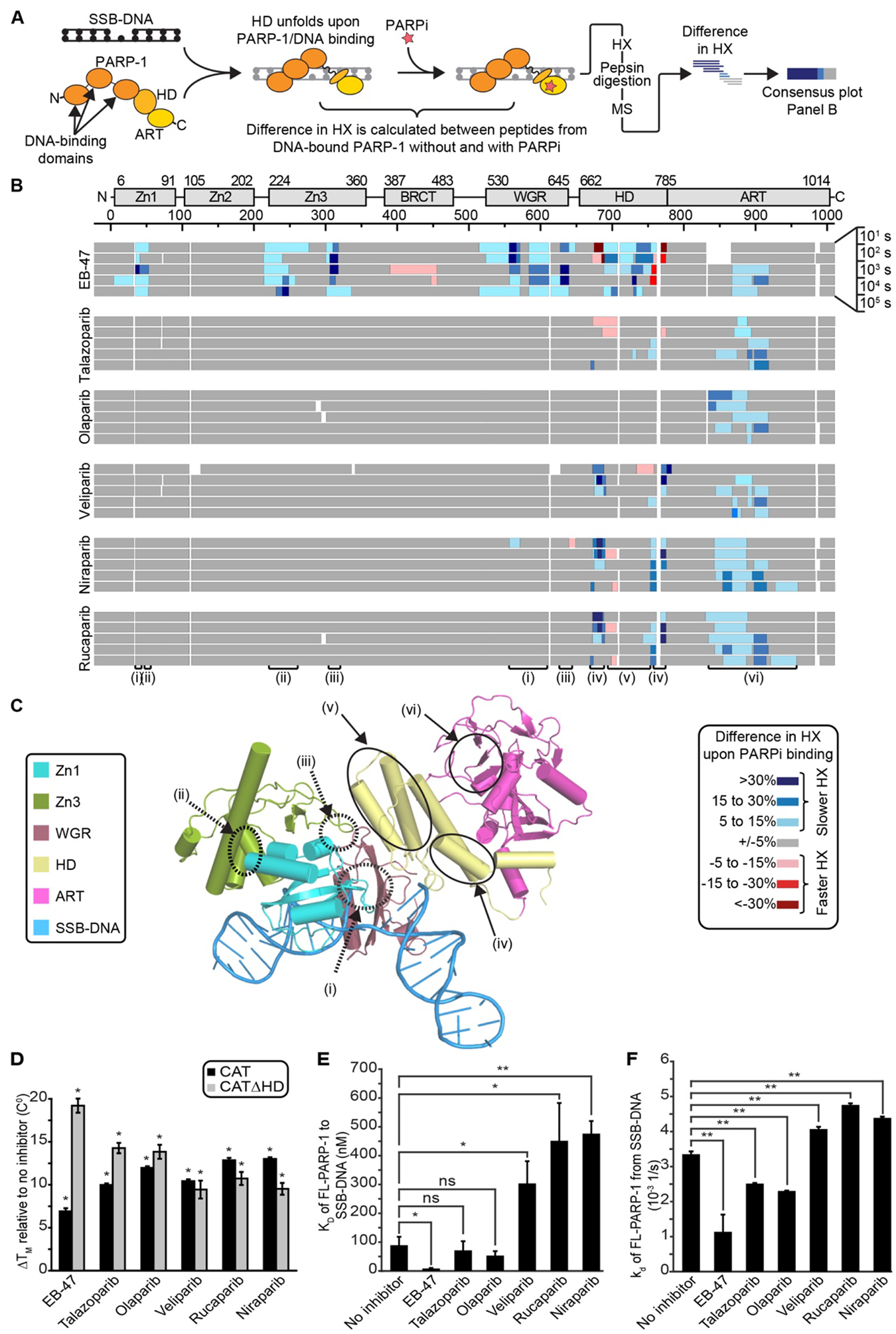
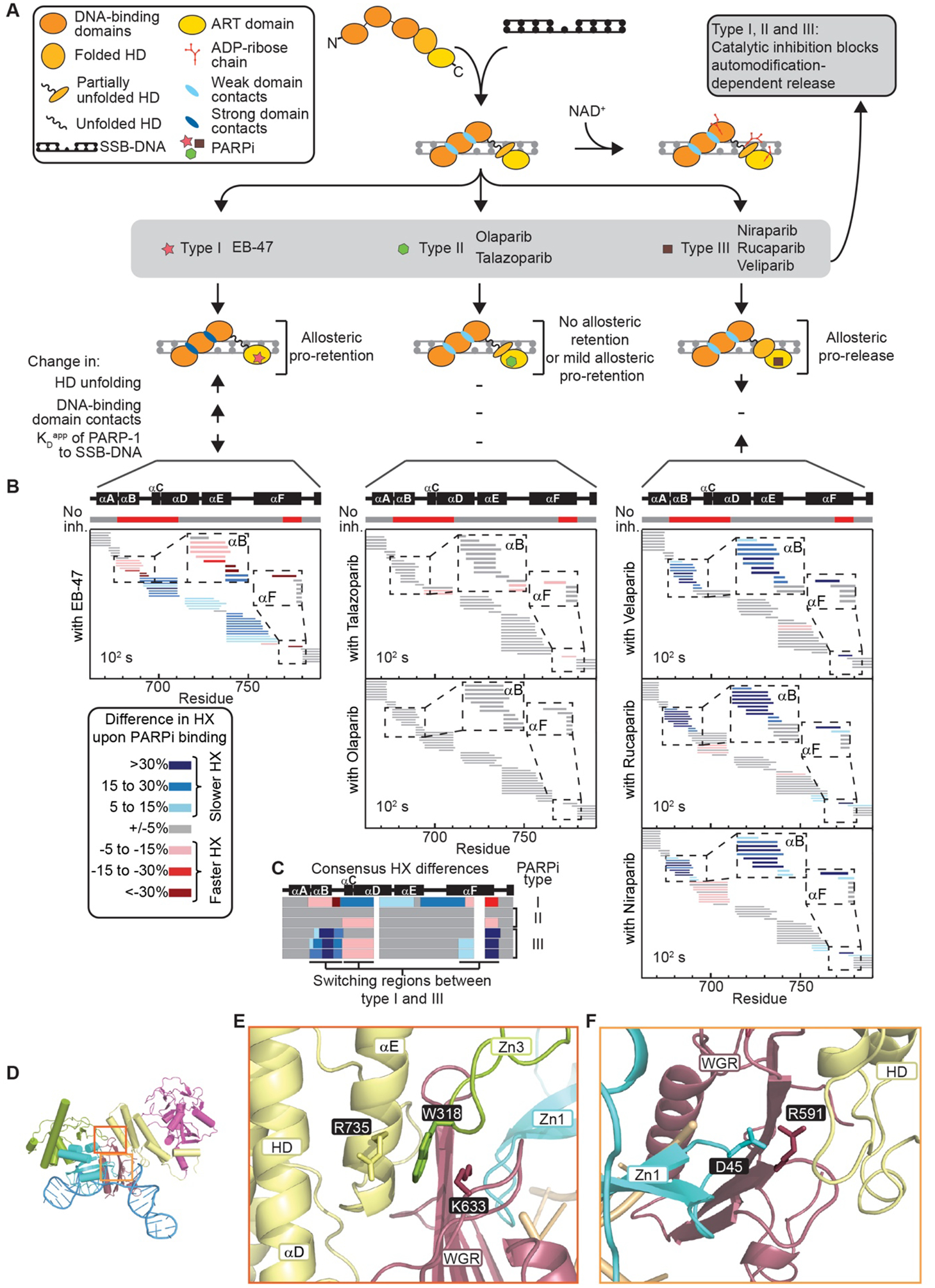
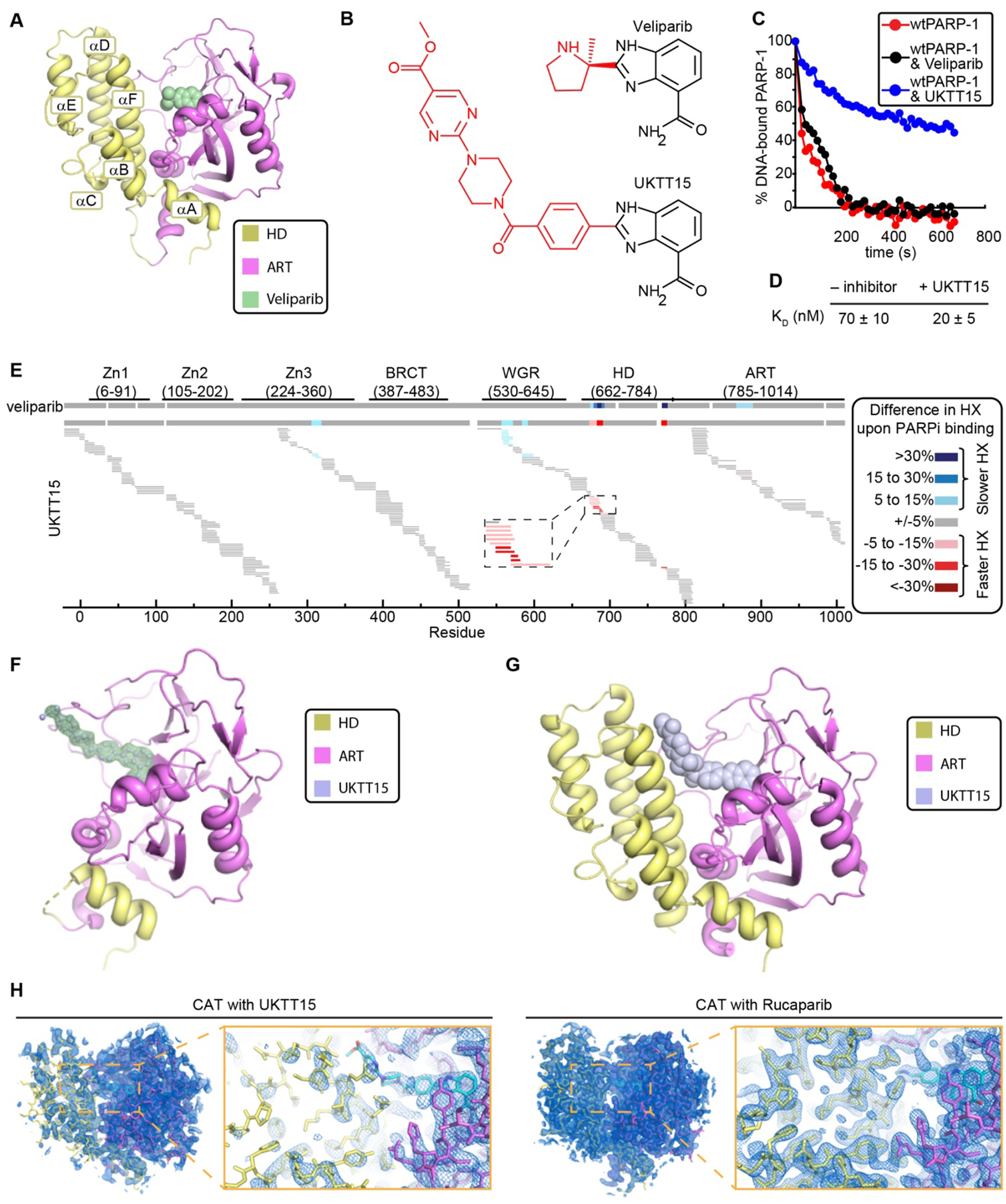
The success of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors (PARPi) to treat cancer relates to their ability to trap PARP-1 at the site of a DNA break. Although different forms of PARPi all target the catalytic center of the enzyme, they have variable abilities to trap PARP-1. We found that several structurally distinct PARPi drive PARP-1 allostery to promote release from a DNA break. Other inhibitors drive allostery to retain PARP-1 on a DNA break. Further, we generated a new PARPi compound, converting an allosteric pro-release compound to a pro-retention compound and increasing its ability to kill cancer cells. These developments are pertinent to clinical applications where PARP-1 trapping is either desirable or undesirable.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures



Comment in
-
Tuning drug binding.Science. 2020 Apr 3;368(6486):30-31. doi: 10.1126/science.abb1462. Science. 2020. PMID: 32241937 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
