

Tumor-penetrating peptide for systemic targeting of Tenascin-C
- PMID: 32242067
- PMCID: PMC7118115
- DOI: 10.1038/s41598-020-62760-y
Tumor-penetrating peptide for systemic targeting of Tenascin-C
Abstract
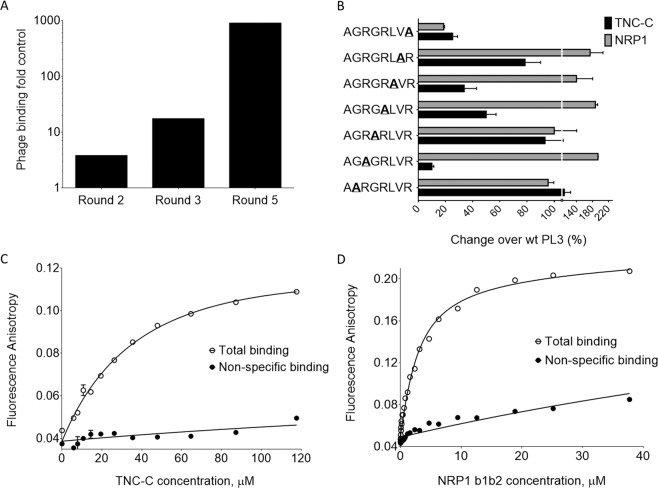
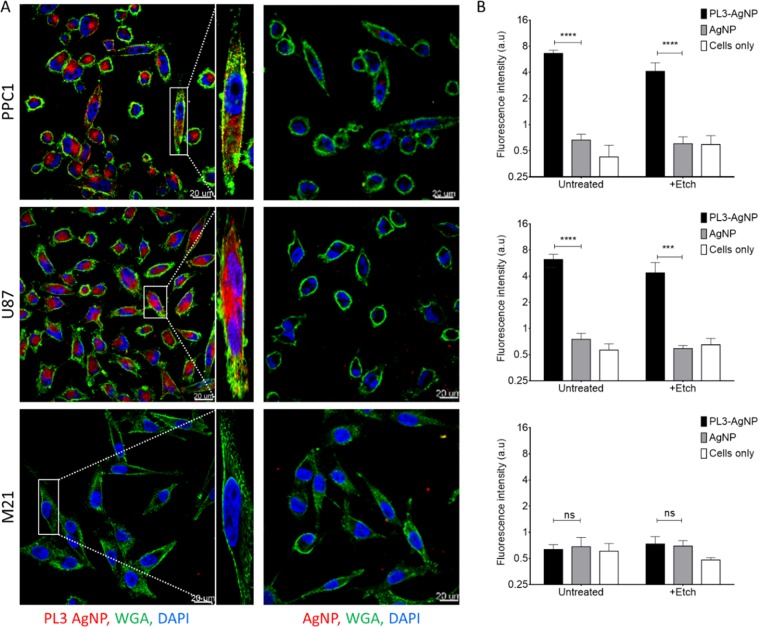
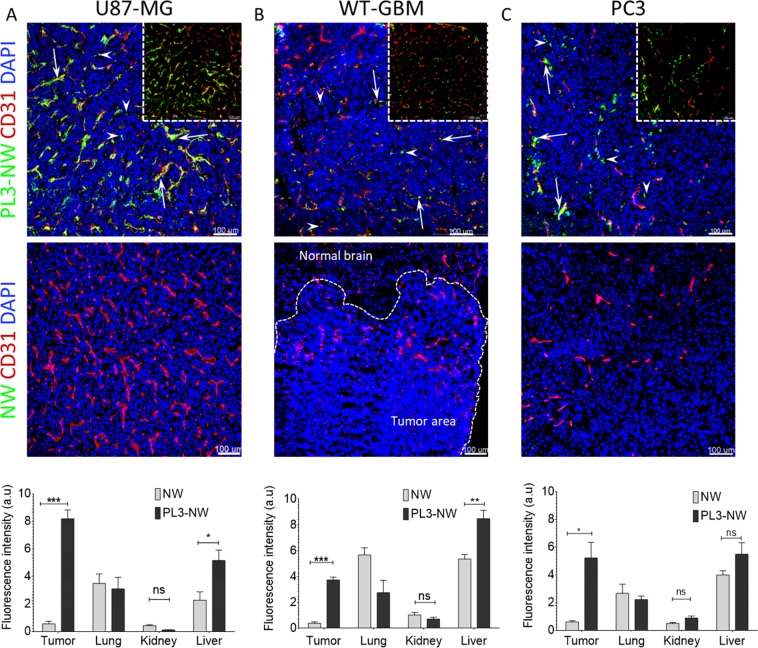
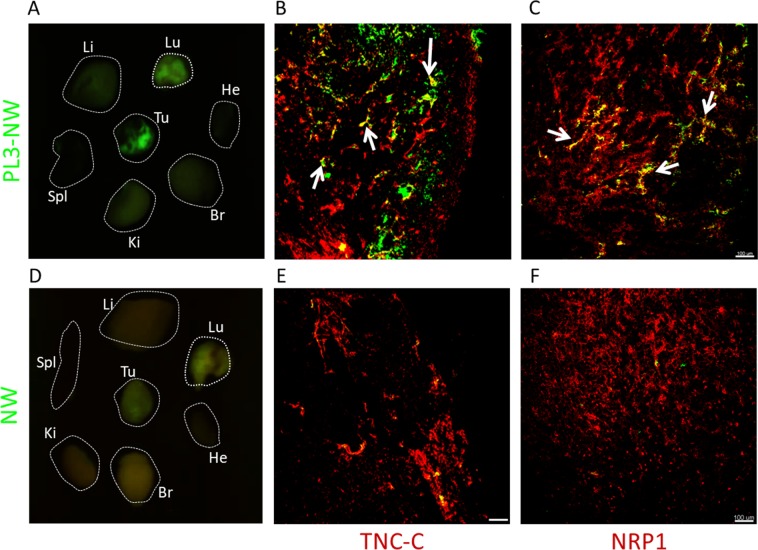
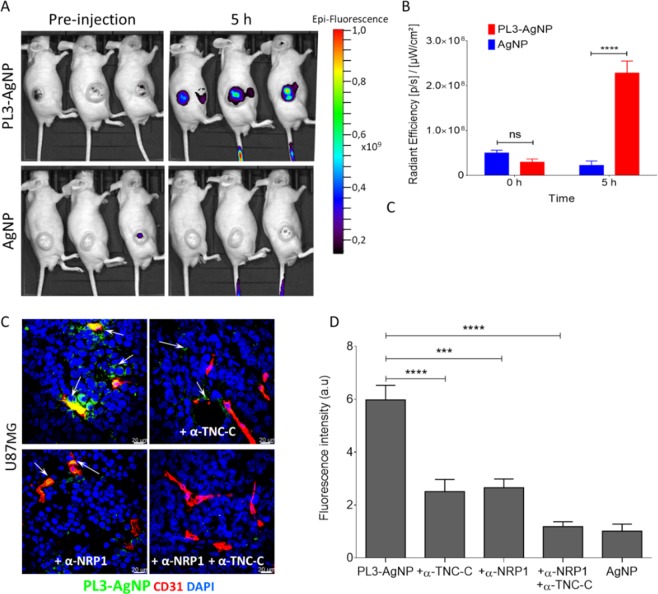
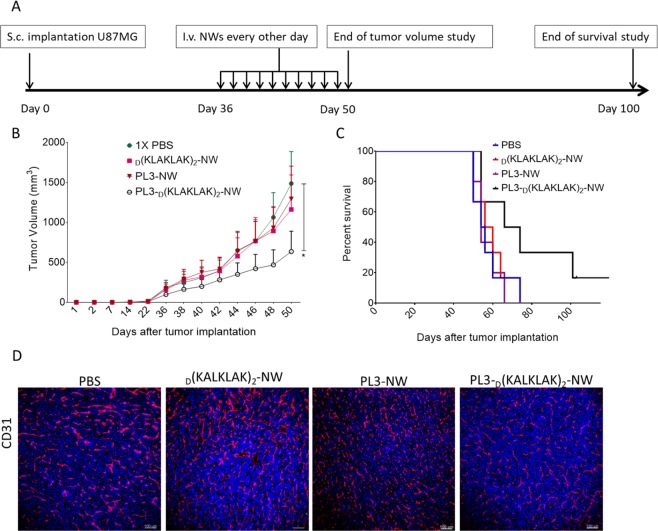
Extracellular matrix in solid tumors has emerged as a specific, stable, and abundant target for affinity-guided delivery of anticancer drugs. Here we describe the homing peptide that interacts with the C-isoform of Tenascin-C (TNC-C) upregulated in malignant tissues. TNC-C binding PL3 peptide (amino acid sequence: AGRGRLVR) was identified by in vitro biopanning on recombinant TNC-C. Besides TNC-C, PL3 interacts via its C-end Rule (CendR) motif with cell-and tissue penetration receptor neuropilin-1 (NRP-1). Functionalization of iron oxide nanoworms (NWs) and metallic silver nanoparticles (AgNPs) with PL3 peptide increased tropism of systemic nanoparticles towards glioblastoma (GBM) and prostate carcinoma xenograft lesions in nude mice (eight and five-fold respectively). Treatment of glioma-bearing mice with proapoptotic PL3-guided NWs improved the survival of the mice, whereas treatment with untargeted particles had no effect. PL3-coated nanoparticles were found to accumulate in TNC-C and NRP-1-positive areas in clinical tumor samples, suggesting a translational relevance. The systemic tumor-targeting properties and binding of PL3-NPs to the clinical tumor sections, suggest that the PL3 peptide may have applications as a targeting moiety for the selective delivery of imaging and therapeutic agents to solid tumors.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
