

Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants
- PMID: 32244552
- PMCID: PMC7231145
- DOI: 10.3390/genes11040378
Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants
Abstract
Aims: We aimed to validate the pathogenicity of genetic variants identified in inherited retinal dystrophy (IRD) patients, which were located in non-canonical splice sites (NCSS).
Methods: After next generation sequencing (NGS) analysis (target gene panels or whole exome sequencing (WES)), NCSS variants were prioritized according to in silico predictions. In vivo and in vitro functional tests were used to validate their pathogenicity.
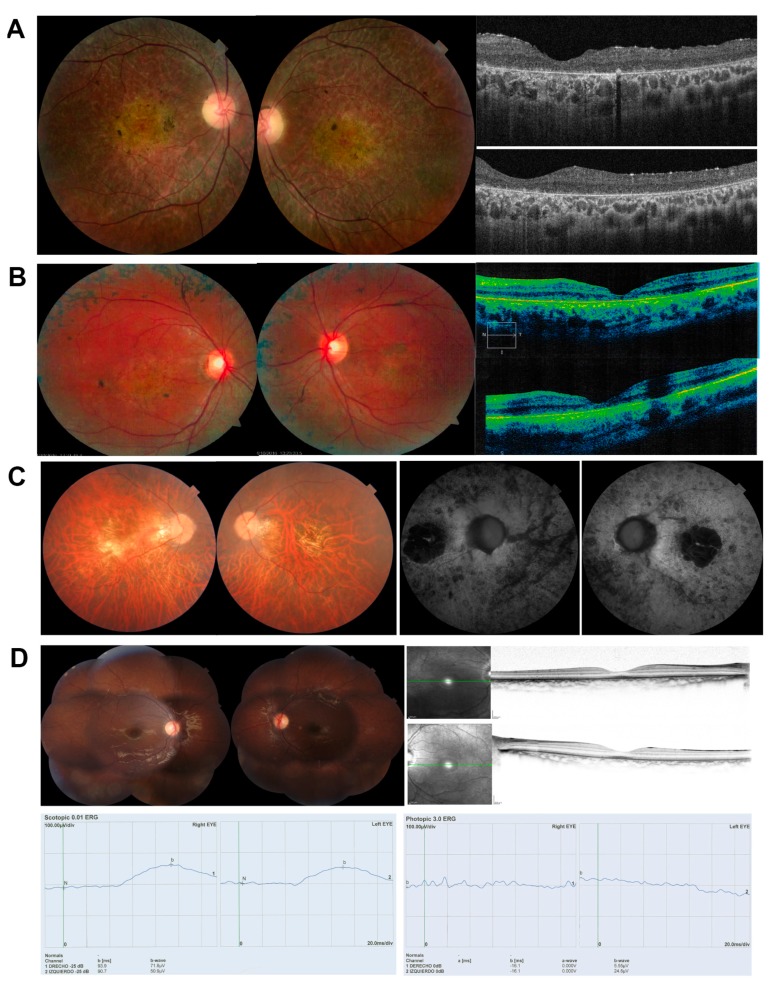
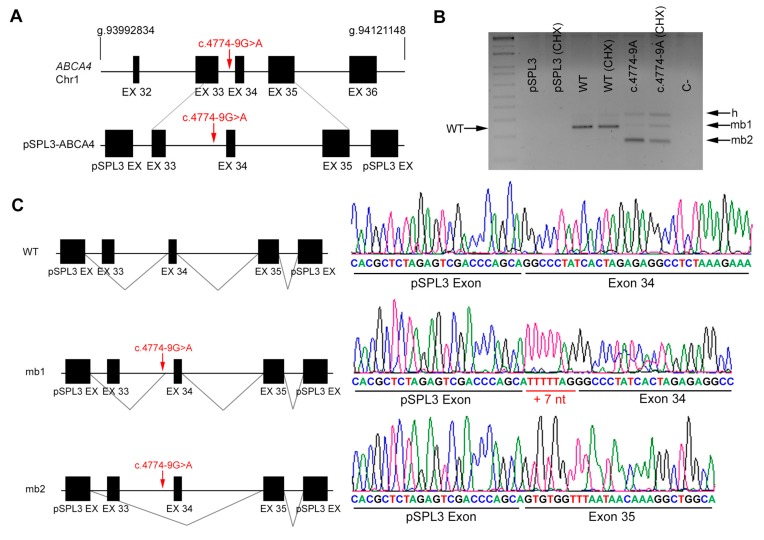
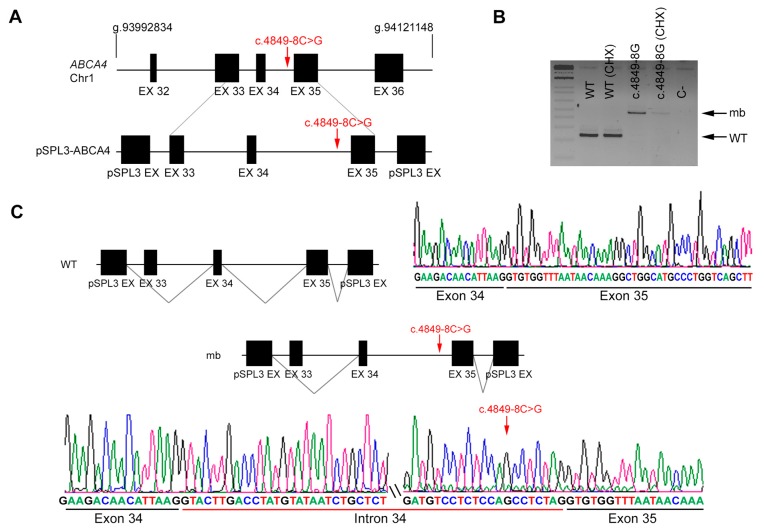
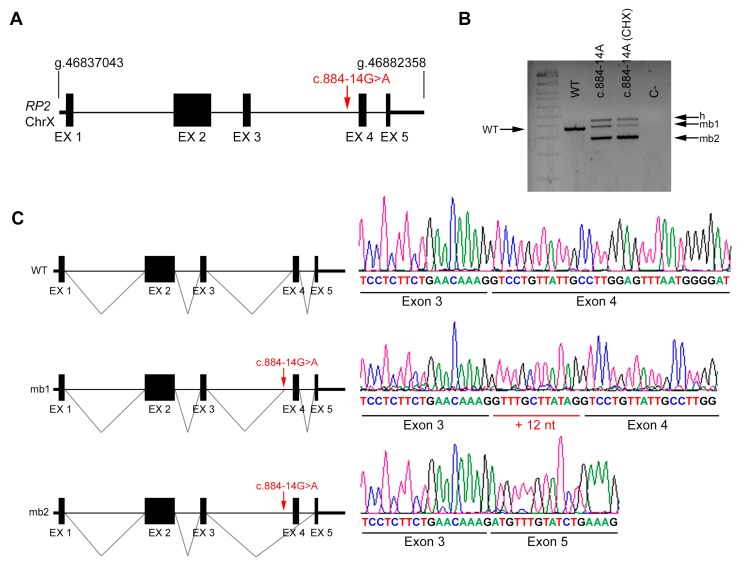
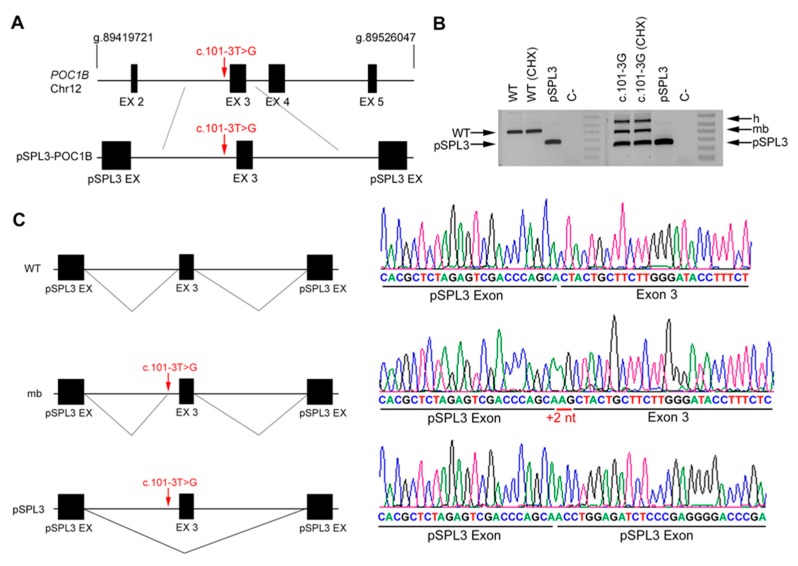
Results: Four novel NCSS variants have been identified. They are located in intron 33 and 34 of ABCA4 (c.4774-9G>A and c.4849-8C>G, respectively), intron 2 of POC1B (c.101-3T>G) and intron 3 of RP2 (c.884-14G>A). Functional analysis detected different aberrant splicing events, including intron retention, exon skipping and intronic nucleotide addition, whose molecular effect was either the disruption or the elongation of the open reading frame of the corresponding gene.
Conclusions: Our data increase the genetic diagnostic yield of IRD patients and expand the landscape of pathogenic variants, which will have an impact on the genotype-phenotype correlations and allow patients to opt for the emerging gene and cell therapies.
Keywords: aberrant splicing; inherited retinal dystrophies; minigenes; non-canonical splice sites.
Conflict of interest statement
The authors hereby declare that there is no competing interest.
Figures
References
-
- Ellingford J.M., Barton S., Bhaskar S., O’Sullivan J., Williams S.G., Lamb J.A., Panda B., Sergouniotis P.I., Gillespie R.L., Daiger S.P., et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016;53:761–767. doi: 10.1136/jmedgenet-2016-103837. - DOI - PMC - PubMed
-
- Sangermano R., Khan M., Cornelis S.S., Richelle V., Albert S., Garanto A., Elmelik D., Qamar R., Lugtenberg D., van den Born L.I., et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018;28:100–110. doi: 10.1101/gr.226621.117. - DOI - PMC - PubMed
-
- Carss K.J., Arno G., Erwood M., Stephens J., Sanchis-Juan A., Hull S., Megy K., Grozeva D., Dewhurst E., Malka S., et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017;100:75–90. doi: 10.1016/j.ajhg.2016.12.003. - DOI - PMC - PubMed
