

Huntingtin Lowering Strategies
- PMID: 32245050
- PMCID: PMC7139361
- DOI: 10.3390/ijms21062146
Huntingtin Lowering Strategies
Abstract
Trials using antisense oligonucleotide technology to lower Huntingtin levels in Huntington's disease (HD) are currently ongoing. This progress, taking place only 27 years after the identification of the Huntingtin gene (HTT) in 1993 reflects the enormous development in genetic engineering in the last decades. It is also the result of passionate basic scientific work and large worldwide registry studies that have advanced the understanding of HD. Increased knowledge of the pathophysiology of this autosomal dominantly inherited CAG-repeat expansion mediated neurodegenerative disease has led to the development of several putative treatment strategies, currently under investigation. These strategies span the whole spectrum of potential targets from genome editing via RNA interference to promoting protein degradation. Yet, recent studies revealed the importance of huntingtin RNA in the pathogenesis of the disease. Therefore, huntingtin-lowering by means of RNA interference appears to be a particular promising strategy. As a matter of fact, these approaches have entered, or are on the verge of entering, the clinical trial period. Here, we provide an overview of huntingtin-lowering approaches via DNA or RNA interference in present clinical trials as well as strategies subject to upcoming therapeutic options. We furthermore discuss putative implications for future treatment of HD patients.
Keywords: Chorea; HD; Huntington’s Disease; RNA interference; antisense oligonucleotides; disease modification; huntingtin-lowering.
Conflict of interest statement
F.M. is Co-PI of Enroll-HD and Generation HD1 at the recruiting site outpatient center for HD, Molecular Neurology, University Hospital Erlangen, Germany.
Figures
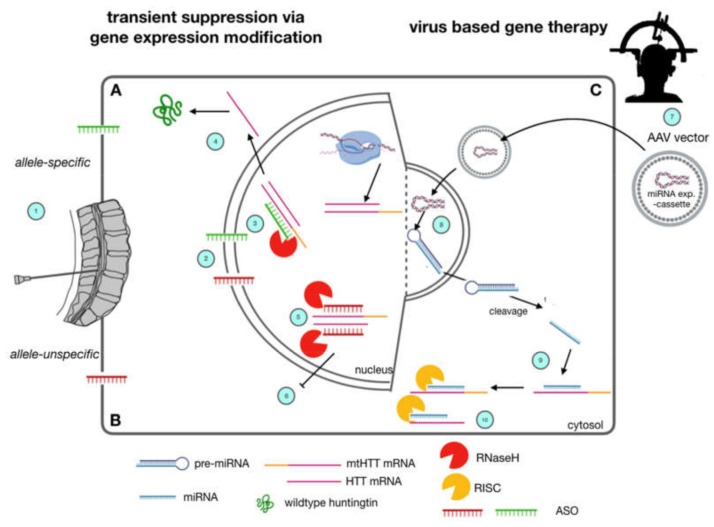
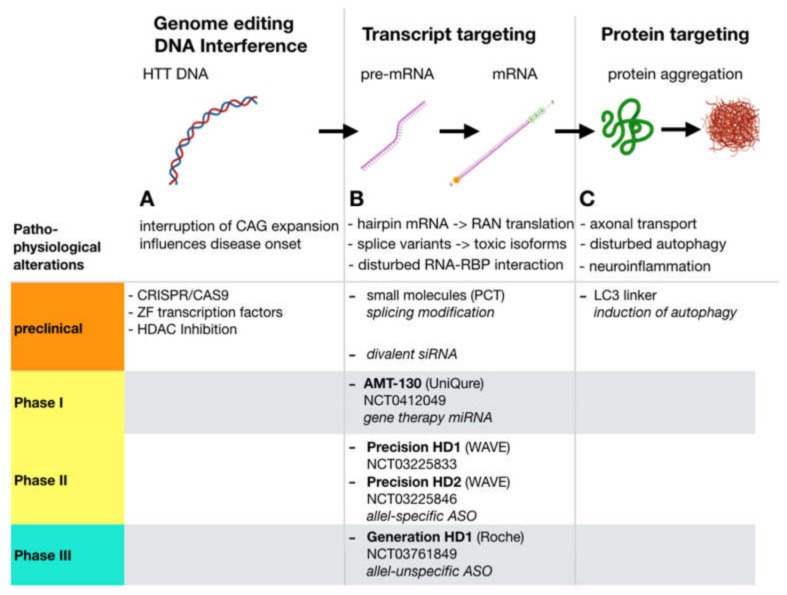
References
-
- Marshall F.J. Tetrabenazine as antichorea therapy in Huntington disease: A randomized controlled trial. Neurology. 2006;66:366–372. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
