

MI-MAAP: marker informativeness for multi-ancestry admixed populations
- PMID: 32245404
- PMCID: PMC7119171
- DOI: 10.1186/s12859-020-3462-5
MI-MAAP: marker informativeness for multi-ancestry admixed populations
Abstract
Background: Admixed populations arise when two or more previously isolated populations interbreed. A powerful approach to addressing the genetic complexity in admixed populations is to infer ancestry. Ancestry inference including the proportion of an individual's genome coming from each population and its ancestral origin along the chromosome of an admixed population requires the use of ancestry informative markers (AIMs) from reference ancestral populations. AIMs exhibit substantial differences in allele frequency between ancestral populations. Given the huge amount of human genetic variation data available from diverse populations, a computationally feasible and cost-effective approach is becoming increasingly important to extract or filter AIMs with the maximum information content for ancestry inference, admixture mapping, forensic applications, and detecting genomic regions that have been under recent selection.
Results: To address this gap, we present MI-MAAP, an easy-to-use web-based bioinformatics tool designed to prioritize informative markers for multi-ancestry admixed populations by utilizing feature selection methods and multiple genomics resources including 1000 Genomes Project and Human Genome Diversity Project. Specifically, this tool implements a novel allele frequency-based feature selection algorithm, Lancaster Estimator of Independence (LEI), as well as other genotype-based methods such as Principal Component Analysis (PCA), Support Vector Machine (SVM), and Random Forest (RF). We demonstrated that MI-MAAP is a useful tool in prioritizing informative markers and accurately classifying ancestral populations. LEI is an efficient feature selection strategy to retrieve ancestry informative variants with different allele frequency/selection pressure among (or between) ancestries without requiring computationally expensive individual-level genotype data.
Conclusions: MI-MAAP has a user-friendly interface which provides researchers an easy and fast way to filter and identify AIMs. MI-MAAP can be accessed at https://research.cchmc.org/mershalab/MI-MAAP/login/.
Keywords: AIMs; Aancestry informative markers; Admixed population; Admixture mapping; LEI; Lancaster estimator of Independence; MI-MAAP.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




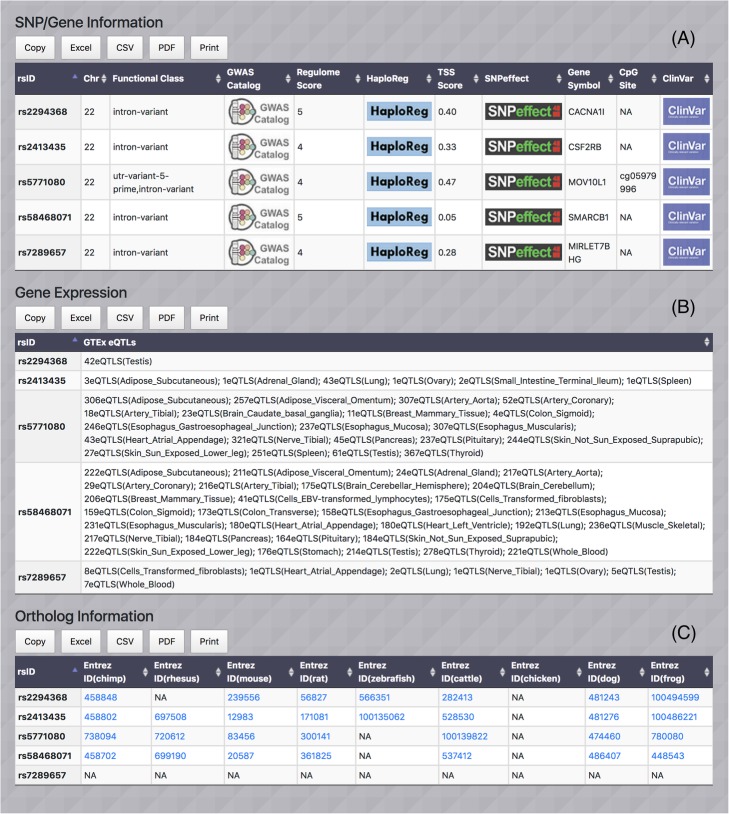

Similar articles
-
LEI: A Novel Allele Frequency-Based Feature Selection Method for Multi-ancestry Admixed Populations.Sci Rep. 2019 Jul 31;9(1):11103. doi: 10.1038/s41598-019-47012-y. Sci Rep. 2019. PMID: 31366927 Free PMC article.
-
An ancestry informative marker panel design for individual ancestry estimation of Hispanic population using whole exome sequencing data.BMC Genomics. 2019 Dec 30;20(Suppl 12):1007. doi: 10.1186/s12864-019-6333-6. BMC Genomics. 2019. PMID: 31888480 Free PMC article.
-
Comparison of measures of marker informativeness for ancestry and admixture mapping.BMC Genomics. 2011 Dec 20;12:622. doi: 10.1186/1471-2164-12-622. BMC Genomics. 2011. PMID: 22185208 Free PMC article.
-
Mapping asthma-associated variants in admixed populations.Front Genet. 2015 Sep 29;6:292. doi: 10.3389/fgene.2015.00292. eCollection 2015. Front Genet. 2015. PMID: 26483834 Free PMC article. Review.
-
Mapping of disease-associated variants in admixed populations.Genome Biol. 2011;12(5):223. doi: 10.1186/gb-2011-12-5-223. Epub 2011 May 30. Genome Biol. 2011. PMID: 21635713 Free PMC article. Review.
Cited by
-
A Pipeline and Recommendations for Population and Individual Diagnostic SNP Selection in Non-Model Species.Mol Ecol Resour. 2025 Apr;25(3):e14048. doi: 10.1111/1755-0998.14048. Epub 2024 Nov 29. Mol Ecol Resour. 2025. PMID: 39611246 Free PMC article.
-
Gene variants, oxidative stress and inflammation in Colombian populations.Biomedica. 2025 May 30;45(2):244-266. doi: 10.7705/biomedica.7220. Biomedica. 2025. PMID: 40493827 Free PMC article. English, Spanish.
-
Fully exploiting SNP arrays: a systematic review on the tools to extract underlying genomic structure.Brief Bioinform. 2022 Mar 10;23(2):bbac043. doi: 10.1093/bib/bbac043. Brief Bioinform. 2022. PMID: 35211719 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
