

Surfactant Protein-A Protects against IL-13-Induced Inflammation in Asthma
- PMID: 32245819
- PMCID: PMC7304346
- DOI: 10.4049/jimmunol.1901227
Surfactant Protein-A Protects against IL-13-Induced Inflammation in Asthma
Abstract
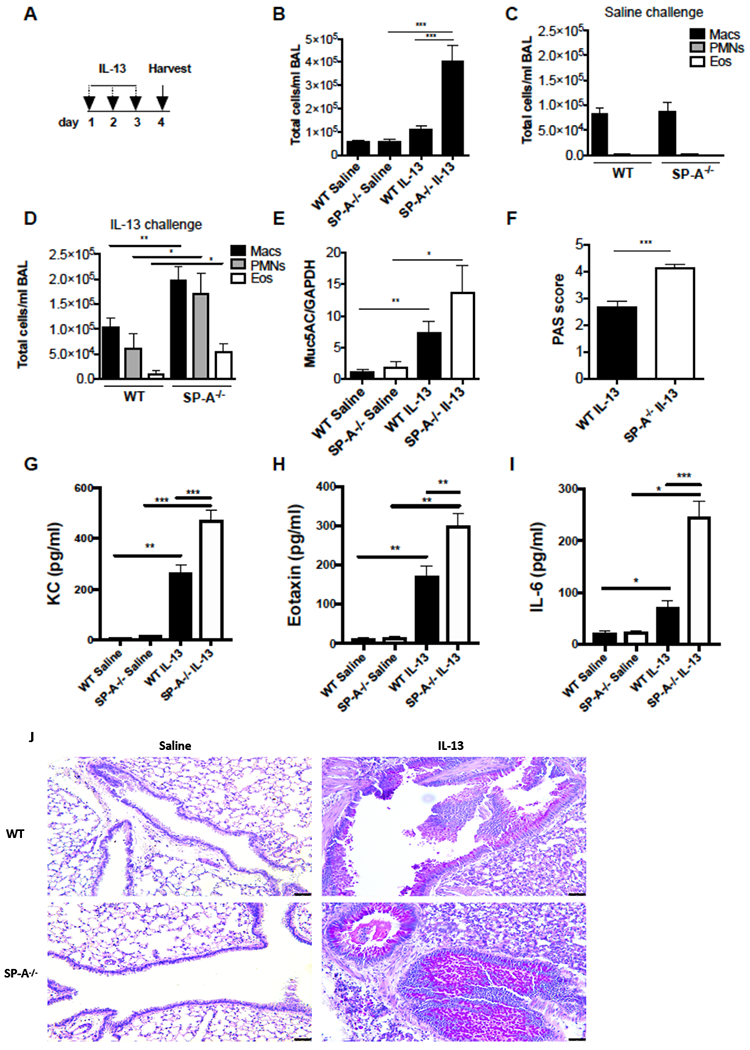
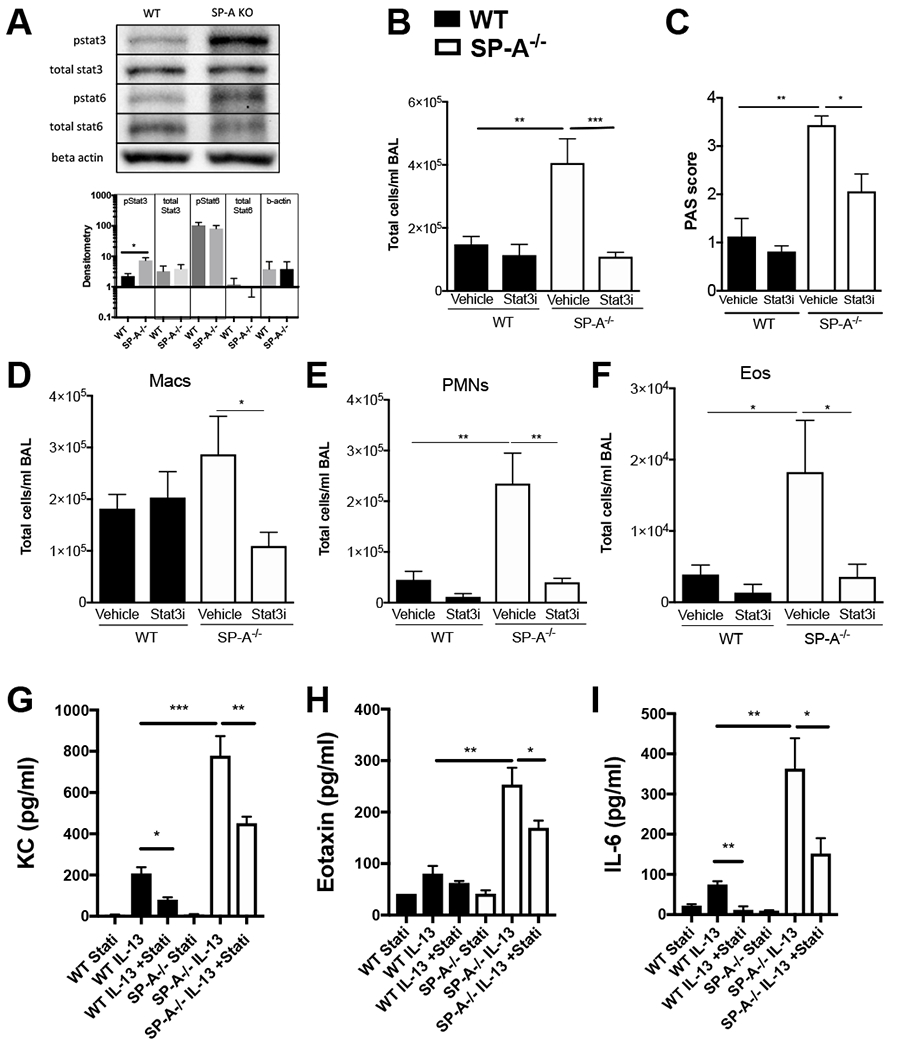
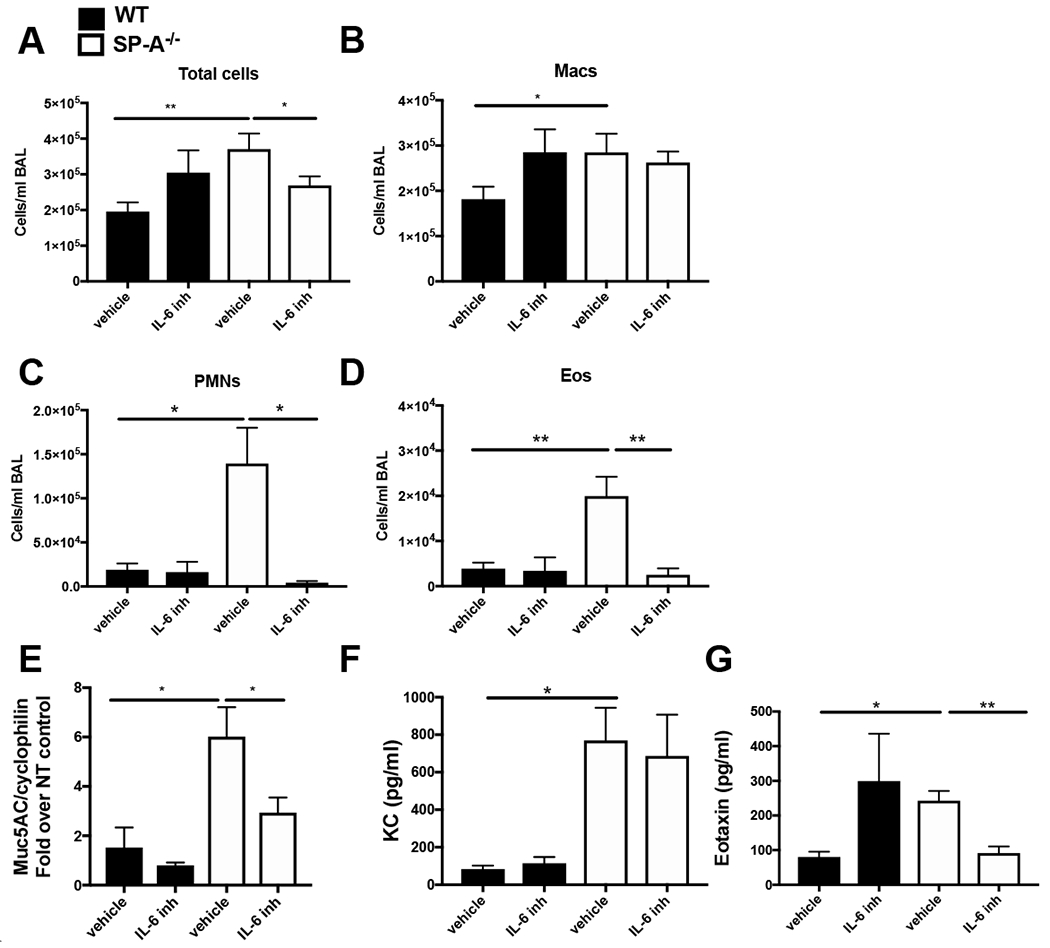
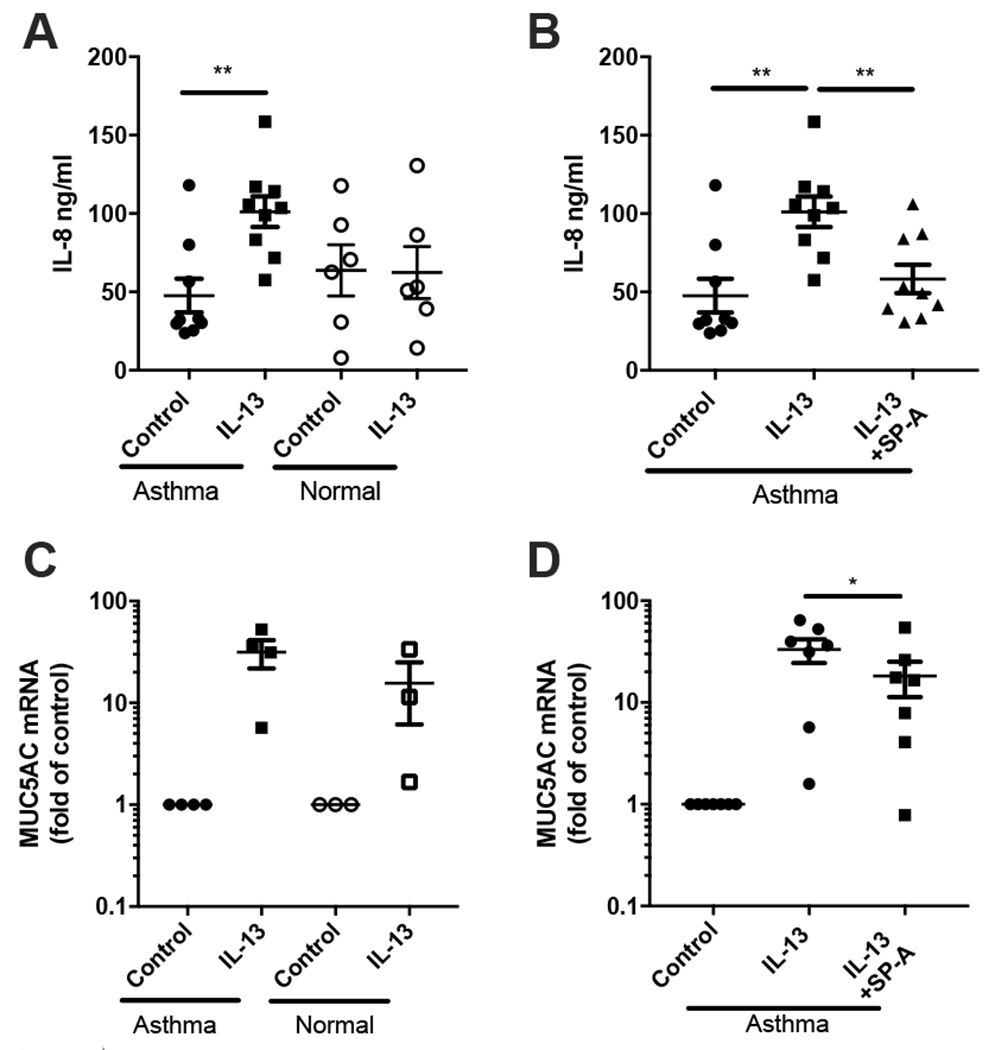
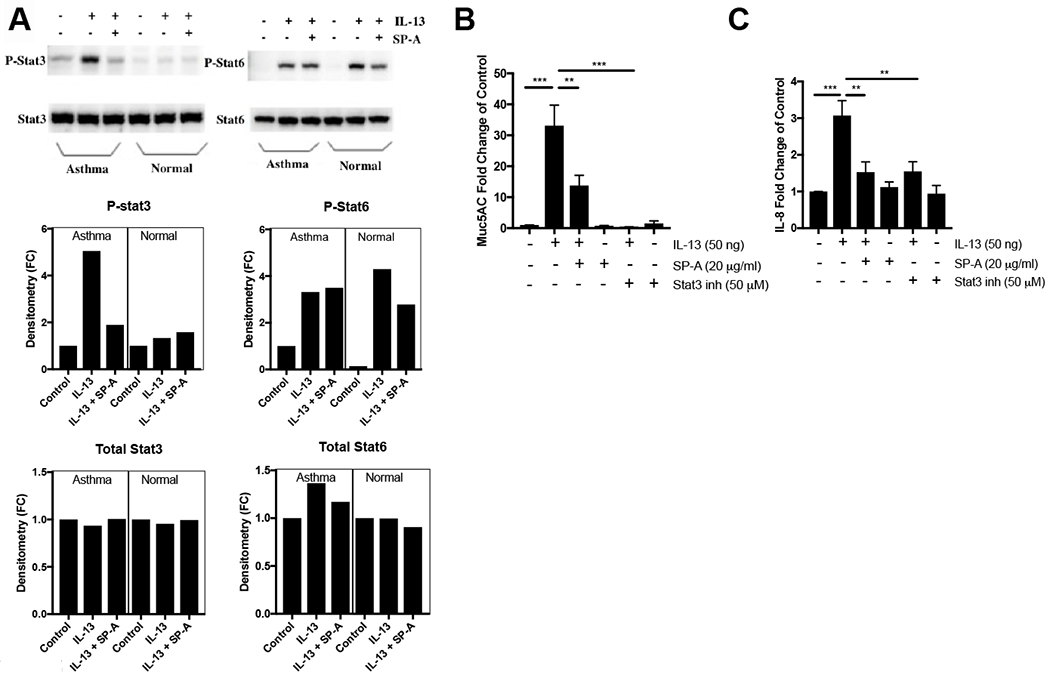
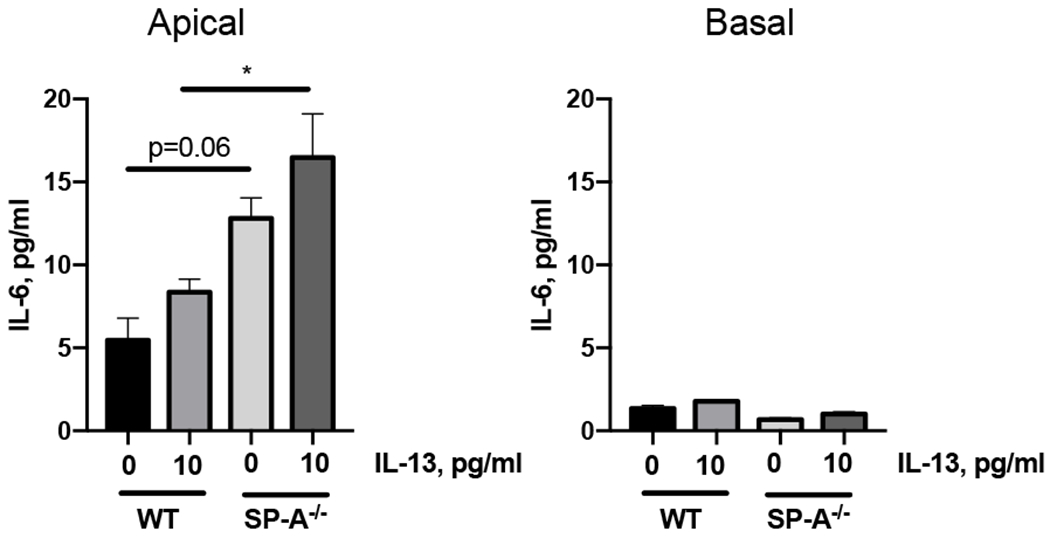
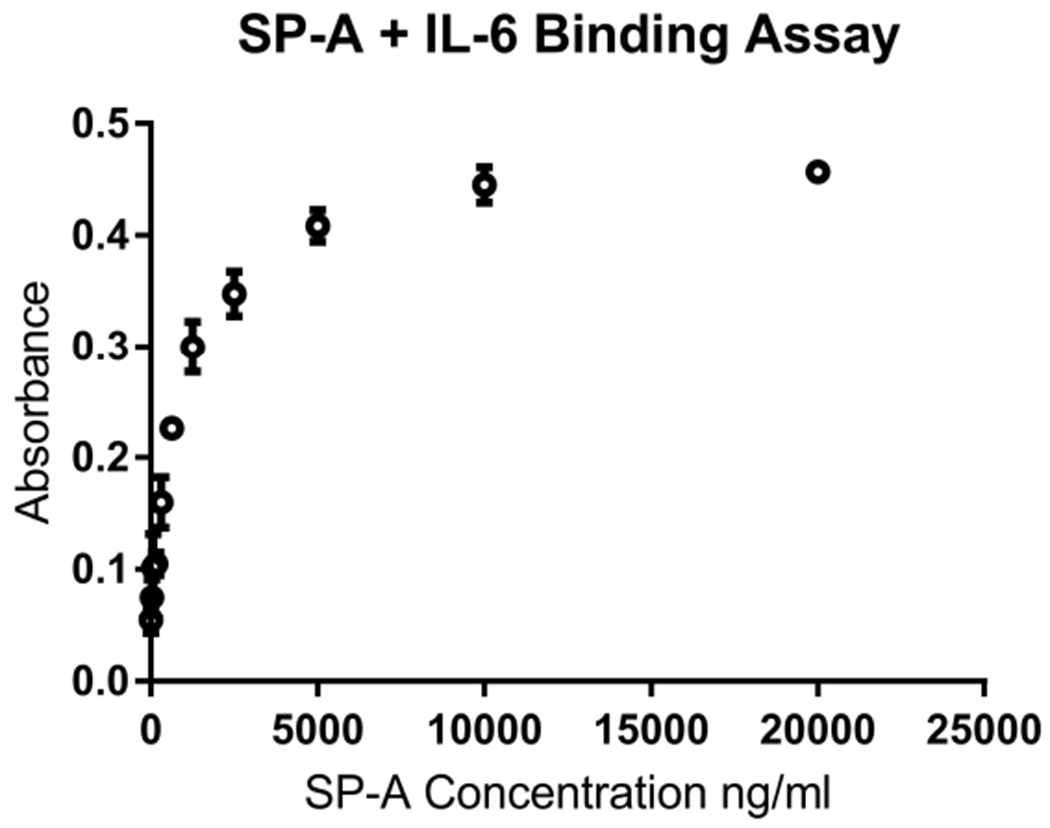
The lung surfactant proteins are recognized as critical not only for their role in lowering lung surface tension but also in innate host defense. Reports have shown that some asthmatic patients have decreased levels of one member of this protein family in particular, surfactant protein-A (SP-A). Our studies set out to determine the contribution of SP-A to the response of a key effector cytokine in asthma, IL-13. Our studies employ both animal models sufficient and deficient in SP-A challenged with IL-13 and primary epithelial cells from participants with asthma that are exogenously treated with SP-A in the context of IL-13 challenge. The inflammatory response and mucin production were assessed in both model systems. As compared with WT mice, we show that the activity of IL-13 is dramatically augmented in SP-A-/- mice, which have significantly increased neutrophil and eosinophil recruitment, mucin production and asthma-associated cytokines in the bronchoalveolar lavage fluid. In parallel, we show asthma-associated factors are attenuated in human cells from asthma subjects when exogenous SP-A is added during IL-13 challenge. Although many of these phenotypes have previously been associated with STAT6 signaling, SP-A inhibited IL-13-induced STAT3 phosphorylation in mice and in human epithelial cells while having little effect on STAT6 phosphorylation. In addition, when either STAT3 or IL-6 were inhibited in mice, the phenotypes observed in SP-A-/- mice were significantly attenuated. These studies suggest a novel mechanism for SP-A in asthma as a modulator of IL-13-induced inflammation via mediating downstream IL-6/STAT3 signaling.
Copyright © 2020 by The American Association of Immunologists, Inc.
Figures
References
-
- Peat JK, Woolcock AJ, and Cullen K. 1987. Rate of decline of lung function in subjects with asthma. Eur J Respir Dis 70: 171–179. - PubMed
-
- Mannino DM, Homa DM, Akinbami LJ, Moorman JE, Gwynn C, and Redd SC. 2002. Surveillance for asthma--United States, 1980–1999. MMWR Surveill Summ 51: 1–13. - PubMed
-
- Djukanovic R, Wilson JW, Britten KM, Wilson SJ, Walls AF, Roche WR, Howarth PH, and Holgate ST. 1992. Effect of an inhaled corticosteroid on airway inflammation and symptoms in asthma. The American review of respiratory disease 145: 669–674. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
