

GenMap: ultra-fast computation of genome mappability
- PMID: 32246826
- PMCID: PMC7320602
- DOI: 10.1093/bioinformatics/btaa222
GenMap: ultra-fast computation of genome mappability
Abstract
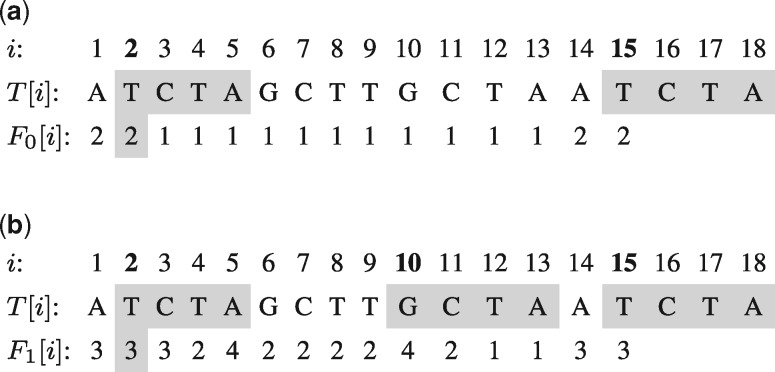
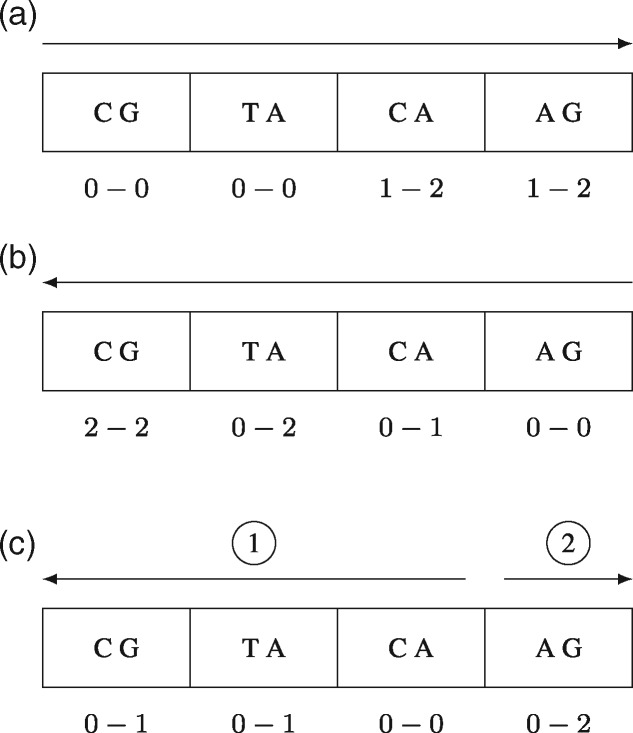
Motivation: Computing the uniqueness of k-mers for each position of a genome while allowing for up to e mismatches is computationally challenging. However, it is crucial for many biological applications such as the design of guide RNA for CRISPR experiments. More formally, the uniqueness or (k, e)-mappability can be described for every position as the reciprocal value of how often this k-mer occurs approximately in the genome, i.e. with up to e mismatches.
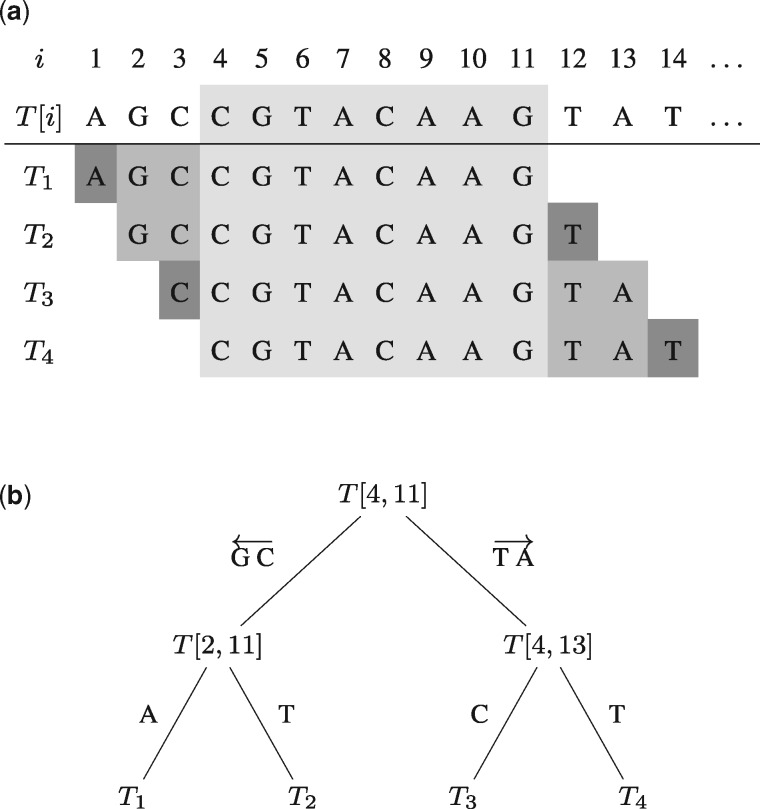
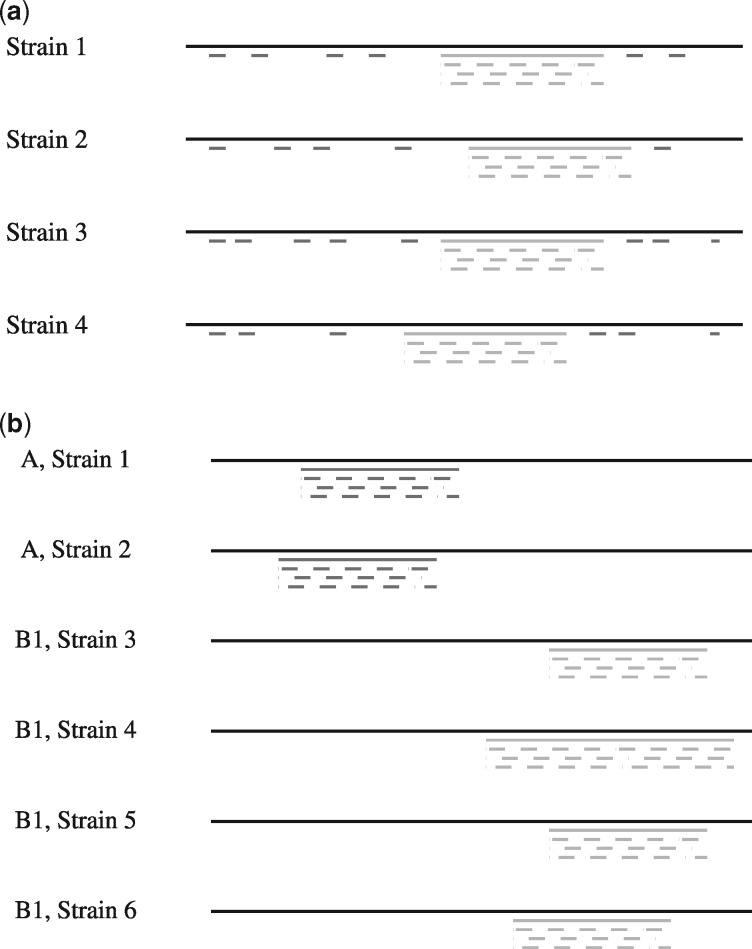
Results: We present a fast method GenMap to compute the (k, e)-mappability. We extend the mappability algorithm, such that it can also be computed across multiple genomes where a k-mer occurrence is only counted once per genome. This allows for the computation of marker sequences or finding candidates for probe design by identifying approximate k-mers that are unique to a genome or that are present in all genomes. GenMap supports different formats such as binary output, wig and bed files as well as csv files to export the location of all approximate k-mers for each genomic position.
Availability and implementation: GenMap can be installed via bioconda. Binaries and C++ source code are available on https://github.com/cpockrandt/genmap.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
-
- Antoniou P. et al. (2009) Mapping uniquely occurring short sequences derived from high throughput technologies to a reference genome In: Information Technology and Applications in Biomedicine (ITAB 2009). IEEE, pp. 1–4. Available at: https://ieeexplore.ieee.org/document/5394394.
-
- Fonseca N.A. et al. (2012) Tools for mapping high-throughput sequencing data. Bioinformatics, 28, 3169–3177. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
