

Missing heritability in Parkinson's disease: the emerging role of non-coding genetic variation
- PMID: 32248367
- PMCID: PMC7242266
- DOI: 10.1007/s00702-020-02184-0
Missing heritability in Parkinson's disease: the emerging role of non-coding genetic variation
Abstract
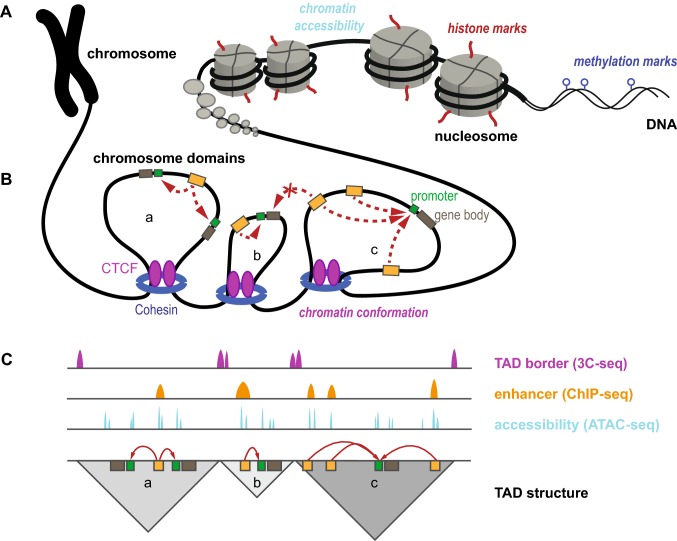
Parkinson's disease (PD) is a neurodegenerative disorder caused by a complex interplay of genetic and environmental factors. For the stratification of PD patients and the development of advanced clinical trials, including causative treatments, a better understanding of the underlying genetic architecture of PD is required. Despite substantial efforts, genome-wide association studies have not been able to explain most of the observed heritability. The majority of PD-associated genetic variants are located in non-coding regions of the genome. A systematic assessment of their functional role is hampered by our incomplete understanding of genotype-phenotype correlations, for example through differential regulation of gene expression. Here, the recent progress and remaining challenges for the elucidation of the role of non-coding genetic variants is reviewed with a focus on PD as a complex disease with multifactorial origins. The function of gene regulatory elements and the impact of non-coding variants on them, and the means to map these elements on a genome-wide level, will be delineated. Moreover, examples of how the integration of functional genomic annotations can serve to identify disease-associated pathways and to prioritize disease- and cell type-specific regulatory variants will be given. Finally, strategies for functional validation and considerations for suitable model systems are outlined. Together this emphasizes the contribution of rare and common genetic variants to the complex pathogenesis of PD and points to remaining challenges for the dissection of genetic complexity that may allow for better stratification, improved diagnostics and more targeted treatments for PD in the future.
Keywords: Gene regulation; Genetic modifier; Genetic susceptibility; Genome-wide association studies; Non-coding variation; Parkinson’s disease; Polygenic risk scores.
Conflict of interest statement
RK serves as Editorial Board Member of the Journal of Neural Transmission, the European Journal of Clinical Investigation and the Journal of Parkinsonism and Related Disorders. RK received speaker’s honoraria and/or travel grants from Abbvie, Zambon and Medtronic and he participated as PI or site-PI for industry-sponsored clinical trials without receiving honoraria. The other authors declare no competing interests.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
