

Transcript, protein, metabolite and cellular studies in skin fibroblasts demonstrate variable pathogenic impacts of NPC1 mutations
- PMID: 32248828
- PMCID: PMC7132889
- DOI: 10.1186/s13023-020-01360-5
Transcript, protein, metabolite and cellular studies in skin fibroblasts demonstrate variable pathogenic impacts of NPC1 mutations
Abstract
Background: Niemann-Pick type C (NP-C) is a rare neurovisceral genetic disorder caused by mutations in the NPC1 or the NPC2 gene. NPC1 is a multipass-transmembrane protein essential for egress of cholesterol from late endosomes/lysosomes. To evaluate impacts of NPC1 mutations, we examined fibroblast cultures from 26 NP-C1 patients with clinical phenotypes ranging from infantile to adult neurologic onset forms. The cells were tested with multiple assays including NPC1 mRNA expression levels and allele expression ratios, assessment of NPC1 promoter haplotypes, NPC1 protein levels, cellular cholesterol staining, localization of the mutant NPC1 proteins to lysosomes, and cholesterol/cholesteryl ester ratios. These results were correlated with phenotypes of the individual patients.
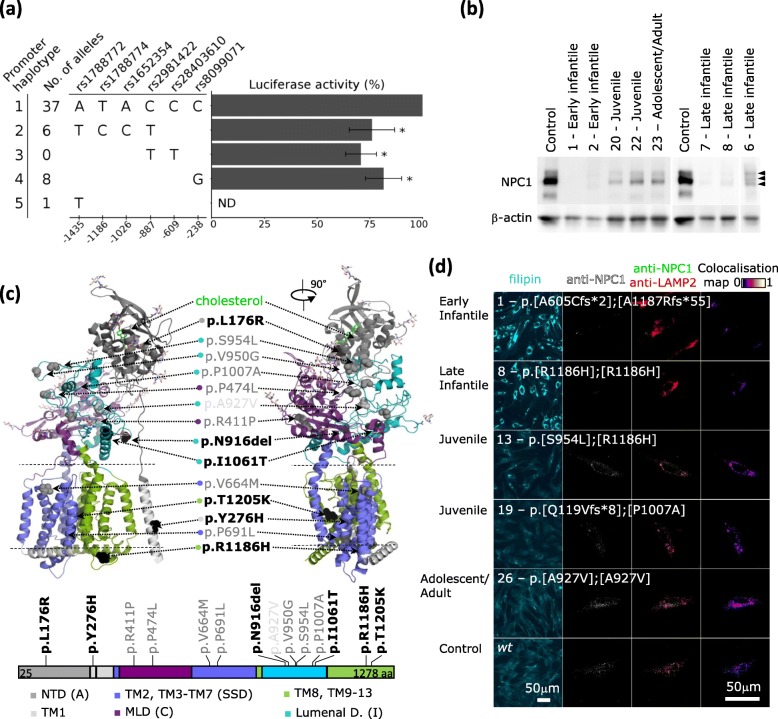
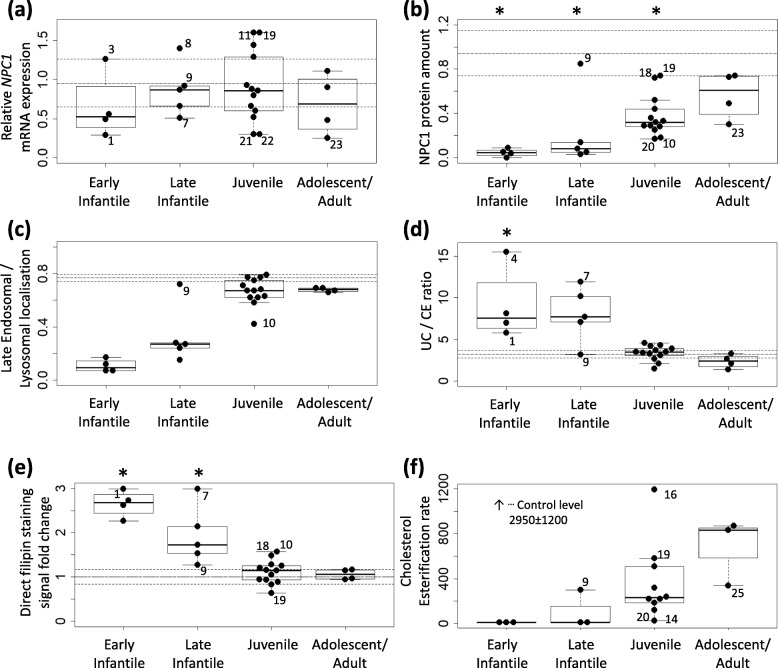
Results: Overall we identified 5 variant promoter haplotypes. Three of them showed reporter activity decreased down to 70% of the control sequence. None of the haplotypes were consistently associated with more severe clinical presentation of NP-C. Levels of transcripts carrying null NPC1 alleles were profoundly lower than levels of the missense variants. Low levels of the mutant NPC1 protein were identified in most samples. The protein localised to lysosomes in cultures expressing medium to normal NPC1 levels. Fibroblasts from patients with severe infantile phenotypes had higher cholesterol levels and higher cholesterol/cholesteryl ester ratios. On the contrary, cell lines from patients with juvenile and adolescent/adult phenotypes showed values comparable to controls.
Conclusion: No single assay fully correlated with the disease severity. However, low residual levels of NPC1 protein and high cholesterol/cholesteryl ester ratios associated with severe disease. The results suggest not only low NPC1 expression due to non-sense mediated decay or low mutant protein stability, but also dysfunction of the stable mutant NPC1 as contributors to the intracellular lipid transport defect.
Keywords: Cholesterol transport; Lysosomal storage disease; Mutant protein; Niemann-pick type C; Proteostasis.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Niemann-Pick C1 functions independently of Niemann-Pick C2 in the initial stage of retrograde transport of membrane-impermeable lysosomal cargo.J Biol Chem. 2010 Feb 12;285(7):4983-94. doi: 10.1074/jbc.M109.037622. Epub 2009 Dec 10. J Biol Chem. 2010. PMID: 20007703 Free PMC article.
-
Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann-pick type C1 disease cells.Hum Mol Genet. 2016 Aug 15;25(16):3588-3599. doi: 10.1093/hmg/ddw204. Epub 2016 Jul 4. Hum Mol Genet. 2016. PMID: 27378690 Free PMC article.
-
Ryanodine receptor antagonists adapt NPC1 proteostasis to ameliorate lipid storage in Niemann-Pick type C disease fibroblasts.Hum Mol Genet. 2012 Jul 15;21(14):3205-14. doi: 10.1093/hmg/dds145. Epub 2012 Apr 14. Hum Mol Genet. 2012. PMID: 22505584 Free PMC article.
-
Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches.J Neurochem. 2011 Mar;116(5):789-95. doi: 10.1111/j.1471-4159.2010.06976.x. Epub 2011 Jan 7. J Neurochem. 2011. PMID: 20807315 Free PMC article. Review.
-
Genetic and laboratory diagnostic approach in Niemann Pick disease type C.J Neurol. 2014 Sep;261 Suppl 2(Suppl 2):S569-75. doi: 10.1007/s00415-014-7386-8. J Neurol. 2014. PMID: 25145893 Free PMC article. Review.
Cited by
-
Skin inflammation and impaired adipogenesis in a mouse model of acid ceramidase deficiency.J Inherit Metab Dis. 2022 Nov;45(6):1175-1190. doi: 10.1002/jimd.12552. Epub 2022 Sep 19. J Inherit Metab Dis. 2022. PMID: 36083604 Free PMC article.
-
Antisense Oligonucleotide-Based Therapy of Viral Infections.Pharmaceutics. 2021 Nov 26;13(12):2015. doi: 10.3390/pharmaceutics13122015. Pharmaceutics. 2021. PMID: 34959297 Free PMC article. Review.
-
At a glance: the largest Niemann-Pick type C1 cohort with 602 patients diagnosed over 15 years.Eur J Hum Genet. 2023 Oct;31(10):1108-1116. doi: 10.1038/s41431-023-01408-7. Epub 2023 Jul 11. Eur J Hum Genet. 2023. PMID: 37433892 Free PMC article.
-
Exploration of Bromodomain Proteins as Drug Targets for Niemann-Pick Type C Disease.Int J Mol Sci. 2025 Jun 16;26(12):5769. doi: 10.3390/ijms26125769. Int J Mol Sci. 2025. PMID: 40565243 Free PMC article.
-
Advances in mass spectrometry of lipids for the investigation of Niemann-pick type C disease.Lipids Health Dis. 2025 Jul 30;24(1):254. doi: 10.1186/s12944-025-02675-7. Lipids Health Dis. 2025. PMID: 40739229 Free PMC article. Review.
References
-
- Jahnova H, Dvorakova L, Vlaskova H, Hulkova H, Poupetova H, Hrebicek M, et al. Observational, retrospective study of a large cohort of patients with Niemann-pick disease type C in the Czech Republic: a surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J Rare Dis. 2014;9:140. doi: 10.1186/s13023-014-0140-6. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
