

Combined Cohesin-RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes
- PMID: 32249213
- PMCID: PMC7269820
- DOI: 10.1158/2159-8290.CD-19-0982
Combined Cohesin-RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes
Abstract
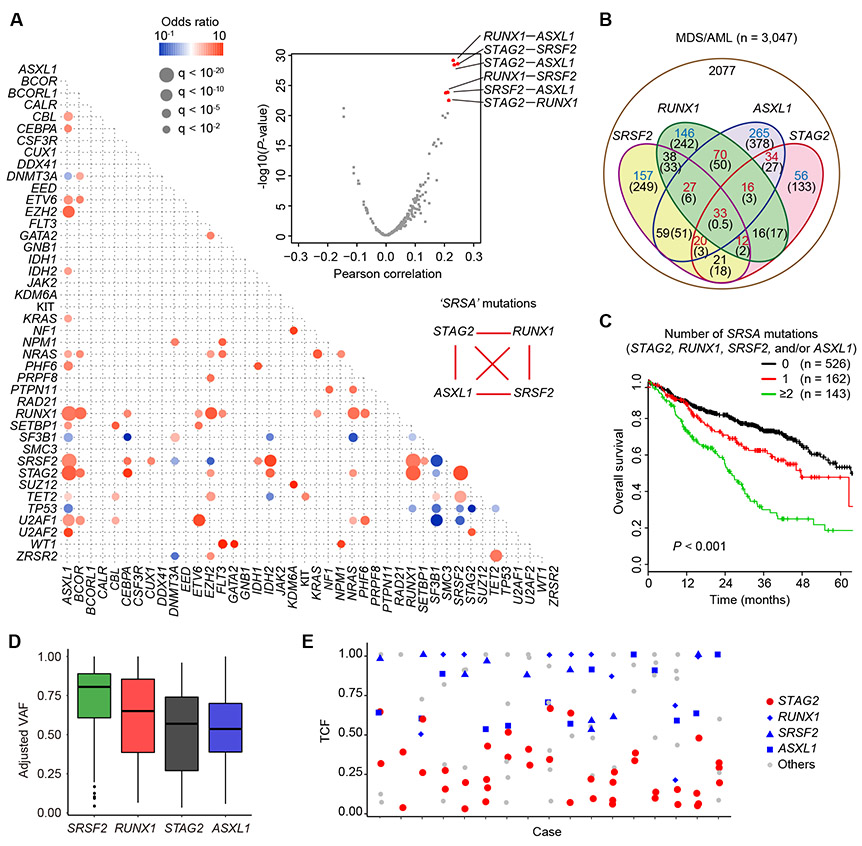
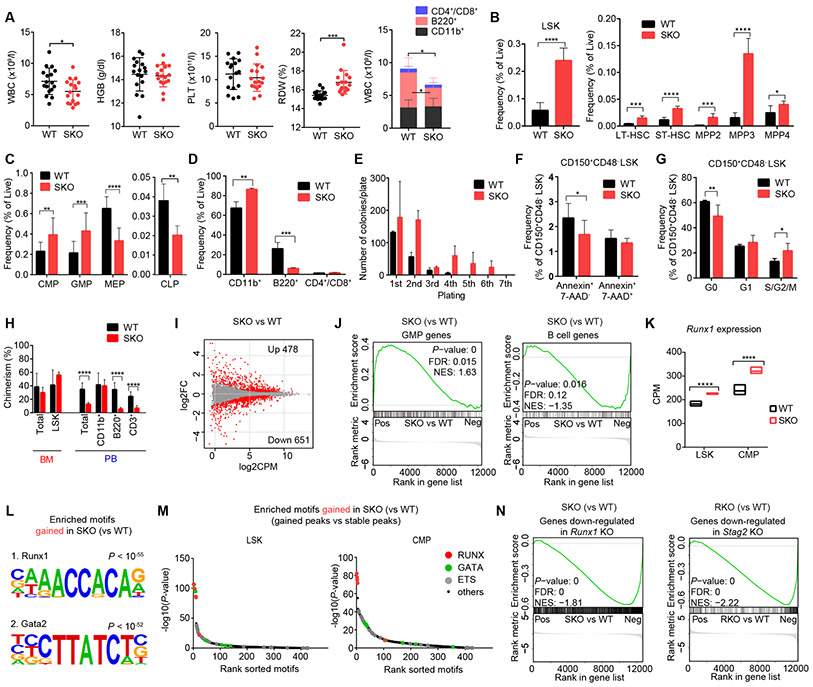
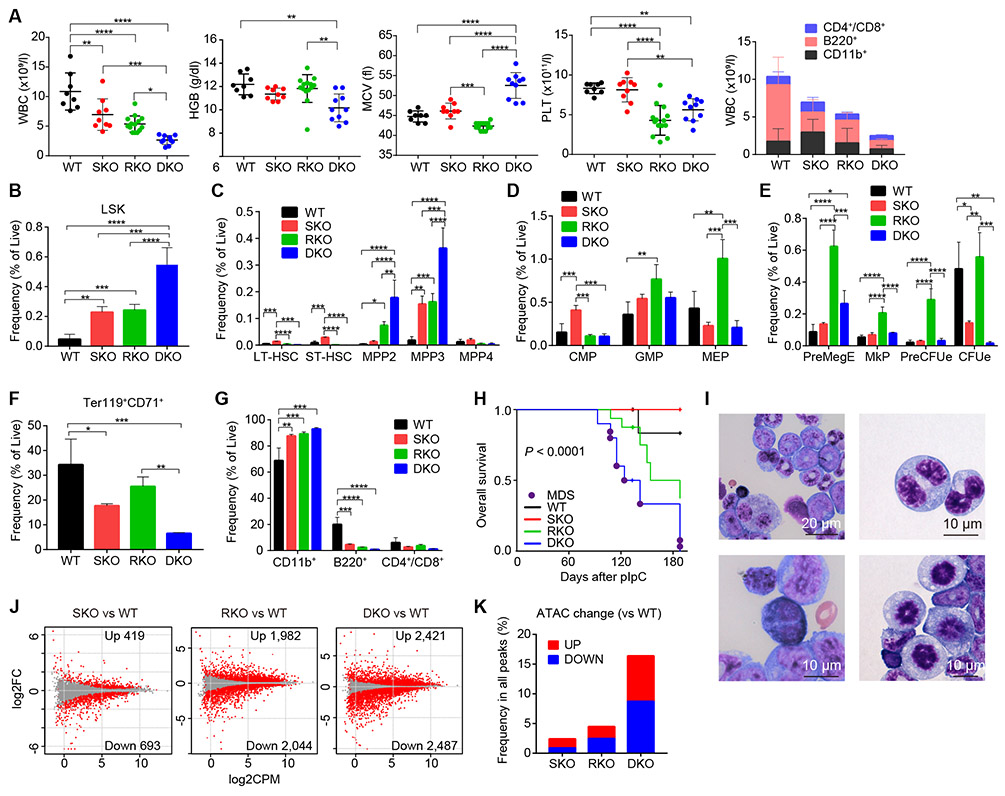
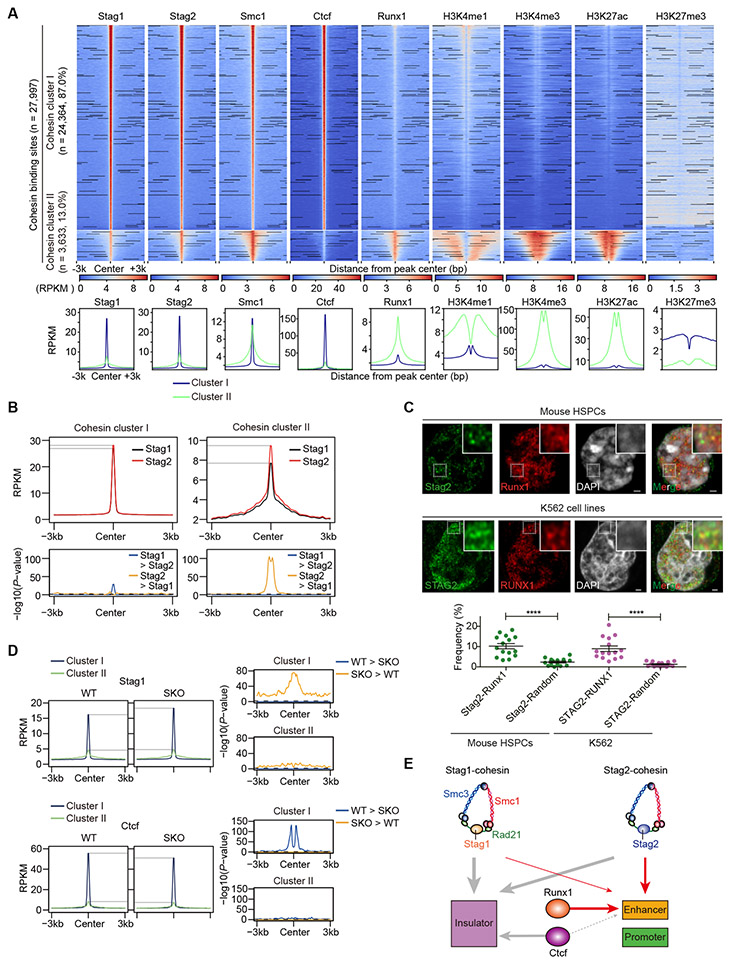
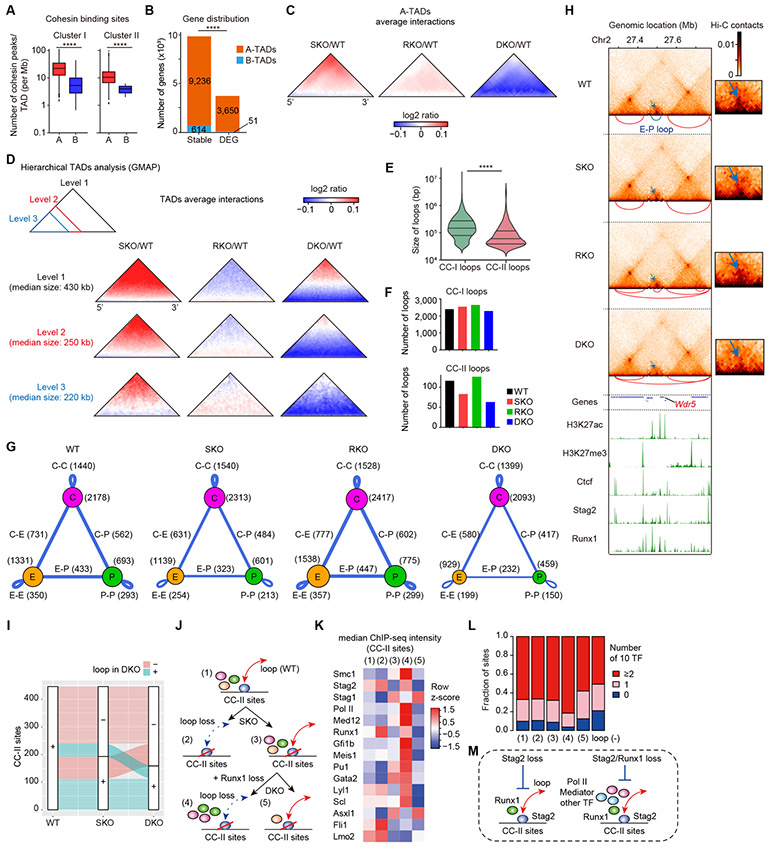
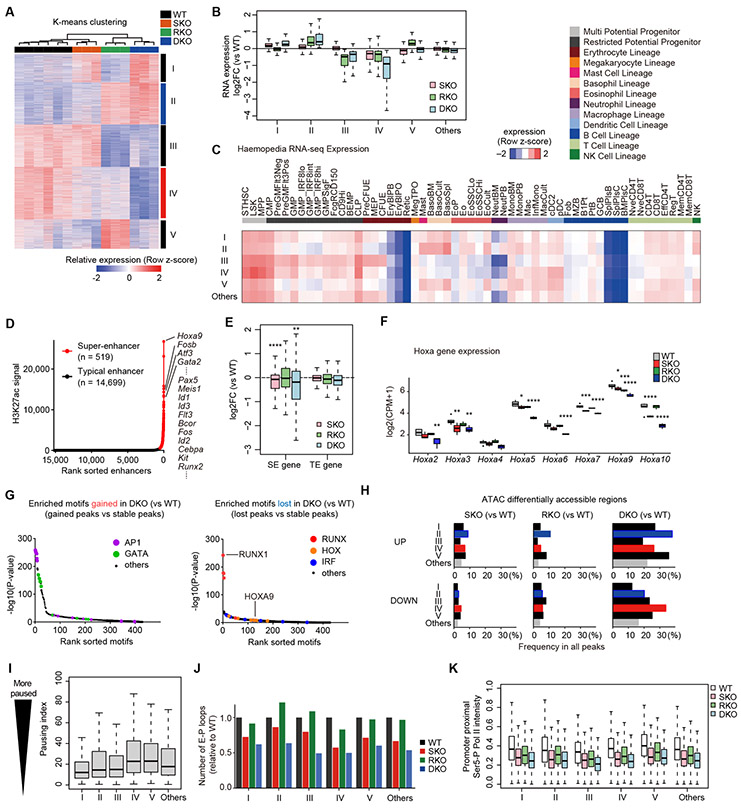
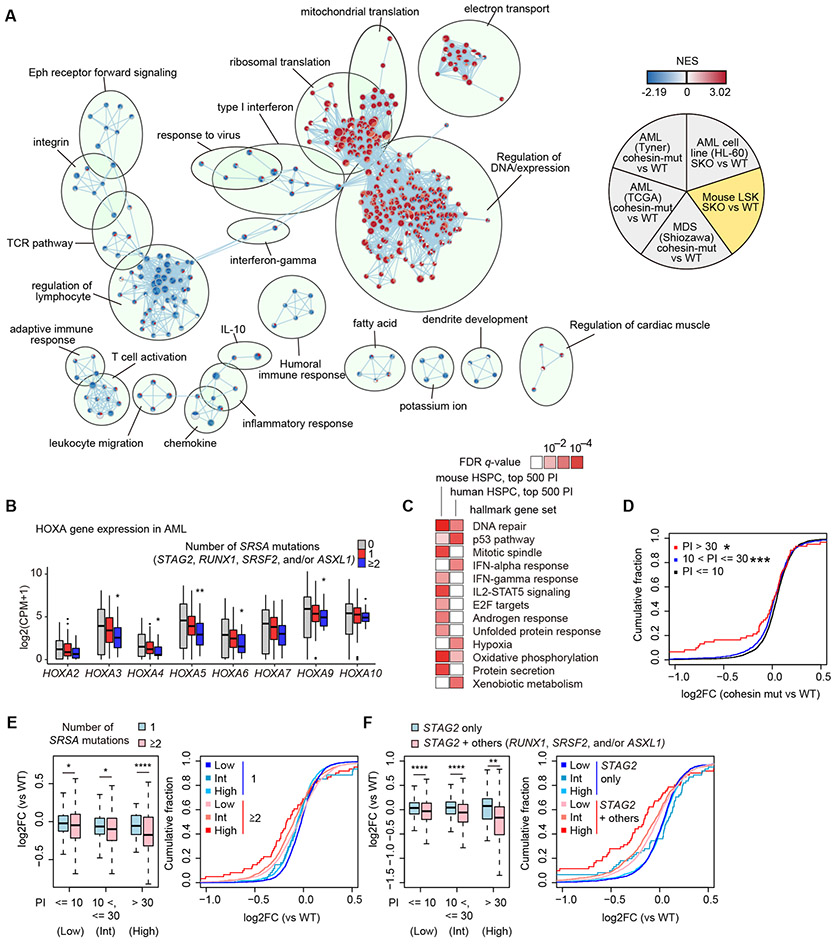
STAG2 encodes a cohesin component and is frequently mutated in myeloid neoplasms, showing highly significant comutation patterns with other drivers, including RUNX1. However, the molecular basis of cohesin-mutated leukemogenesis remains poorly understood. Here we show a critical role of an interplay between STAG2 and RUNX1 in the regulation of enhancer-promoter looping and transcription in hematopoiesis. Combined loss of STAG2 and RUNX1, which colocalize at enhancer-rich, CTCF-deficient sites, synergistically attenuates enhancer-promoter loops, particularly at sites enriched for RNA polymerase II and Mediator, and deregulates gene expression, leading to myeloid-skewed expansion of hematopoietic stem/progenitor cells (HSPC) and myelodysplastic syndromes (MDS) in mice. Attenuated enhancer-promoter loops in STAG2/RUNX1-deficient cells are associated with downregulation of genes with high basal transcriptional pausing, which are important for regulation of HSPCs. Downregulation of high-pausing genes is also confirmed in STAG2-cohesin-mutated primary leukemia samples. Our results highlight a unique STAG2-RUNX1 interplay in gene regulation and provide insights into cohesin-mutated leukemogenesis. SIGNIFICANCE: We demonstrate a critical role of an interplay between STAG2 and a master transcription factor of hematopoiesis, RUNX1, in MDS development, and further reveal their contribution to regulation of high-order chromatin structures, particularly enhancer-promoter looping, and the link between transcriptional pausing and selective gene dysregulation caused by cohesin deficiency.This article is highlighted in the In This Issue feature, p. 747.
©2020 American Association for Cancer Research.
Conflict of interest statement
Conflict of interest disclosure:
The authors declare no conflict of interest.
Figures
Similar articles
-
Untangling the loops of STAG2 mutations in myelodysplastic syndrome.Leuk Lymphoma. 2025 Jan;66(1):6-15. doi: 10.1080/10428194.2024.2400210. Epub 2024 Sep 12. Leuk Lymphoma. 2025. PMID: 39264305 Review.
-
[Molecular pathogenesis of myelodysplastic syndromes with concurrent mutations in cohesin STAG2 and transcription factor RUNX1].Rinsho Ketsueki. 2021;62(5):352-359. doi: 10.11406/rinketsu.62.352. Rinsho Ketsueki. 2021. PMID: 34108313 Japanese.
-
STAG2 mutations reshape the cohesin-structured spatial chromatin architecture to drive gene regulation in acute myeloid leukemia.Cell Rep. 2024 Aug 27;43(8):114498. doi: 10.1016/j.celrep.2024.114498. Epub 2024 Jul 30. Cell Rep. 2024. PMID: 39084219
-
Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation.Cell Stem Cell. 2019 Nov 7;25(5):682-696.e8. doi: 10.1016/j.stem.2019.08.003. Epub 2019 Sep 5. Cell Stem Cell. 2019. PMID: 31495782 Free PMC article.
-
The consequences of cohesin mutations in myeloid malignancies.Front Mol Biosci. 2023 Nov 15;10:1319804. doi: 10.3389/fmolb.2023.1319804. eCollection 2023. Front Mol Biosci. 2023. PMID: 38033389 Free PMC article. Review.
Cited by
-
Molecular and clinical analyses of PHF6 mutant myeloid neoplasia provide their pathogenesis and therapeutic targeting.Nat Commun. 2024 Feb 28;15(1):1832. doi: 10.1038/s41467-024-46134-w. Nat Commun. 2024. PMID: 38418452 Free PMC article.
-
Cohesin mutations in myeloid malignancies.Blood. 2021 Aug 26;138(8):649-661. doi: 10.1182/blood.2019004259. Blood. 2021. PMID: 34157074 Free PMC article. Review.
-
Untangling the loops of STAG2 mutations in myelodysplastic syndrome.Leuk Lymphoma. 2025 Jan;66(1):6-15. doi: 10.1080/10428194.2024.2400210. Epub 2024 Sep 12. Leuk Lymphoma. 2025. PMID: 39264305 Review.
-
Cohesin composition and dosage independently affect early development in zebrafish.Development. 2024 Aug 1;151(15):dev202593. doi: 10.1242/dev.202593. Epub 2024 Aug 1. Development. 2024. PMID: 38975838 Free PMC article.
-
Chromatin organization in myelodysplastic syndrome.Exp Hematol. 2024 Jun;134:104216. doi: 10.1016/j.exphem.2024.104216. Epub 2024 Apr 4. Exp Hematol. 2024. PMID: 38582293 Free PMC article. Review.
References
-
- Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013;45:1232–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
