

Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model
- PMID: 32250339
- PMCID: PMC7190940
- DOI: 10.1172/JCI133316
Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model
Abstract
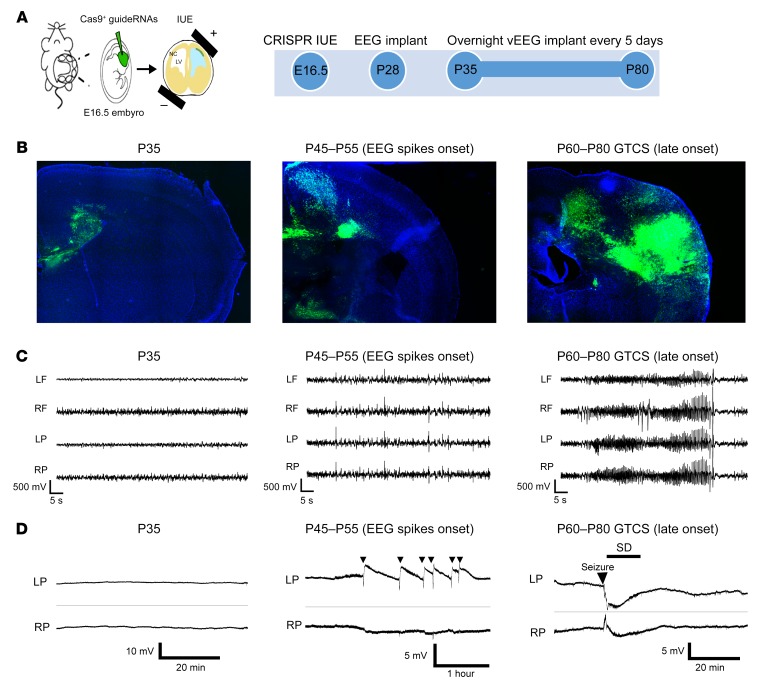
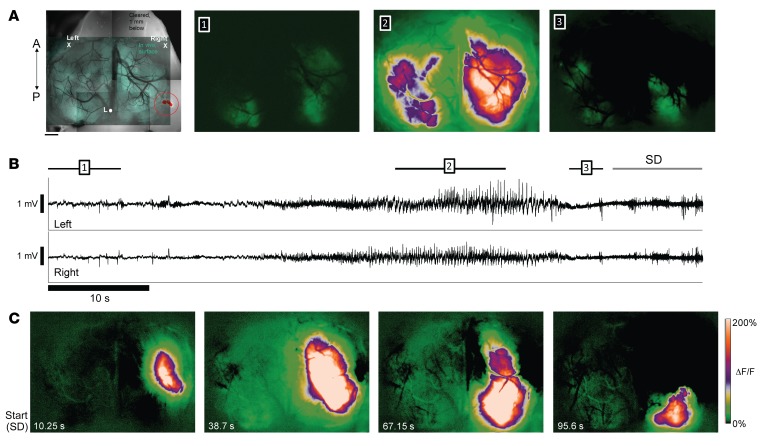
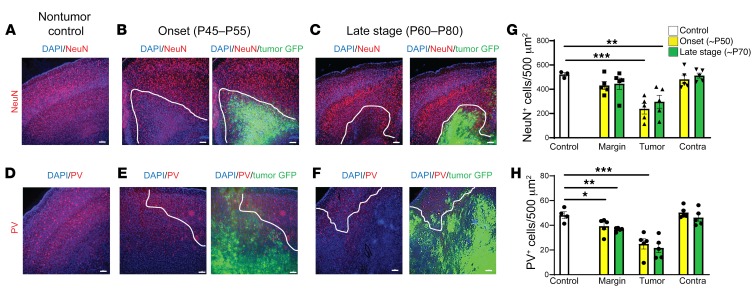
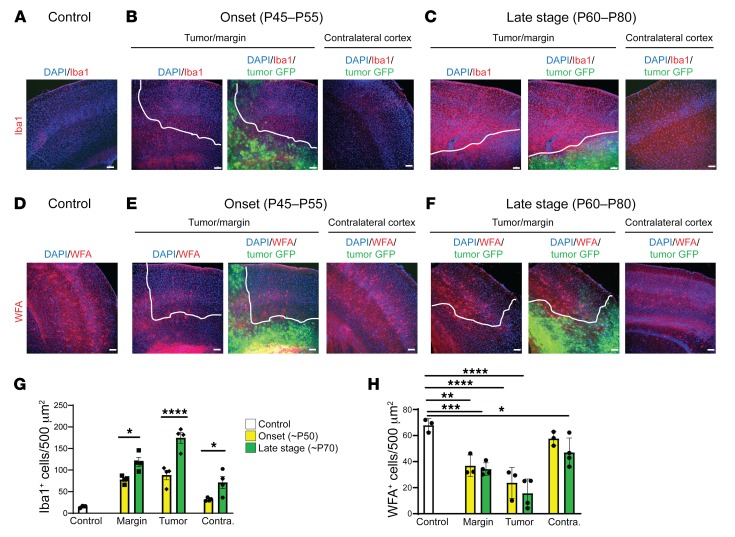
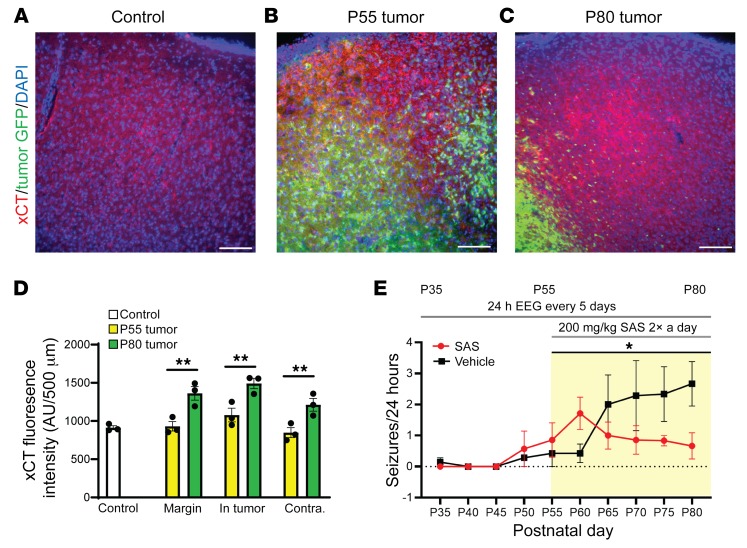
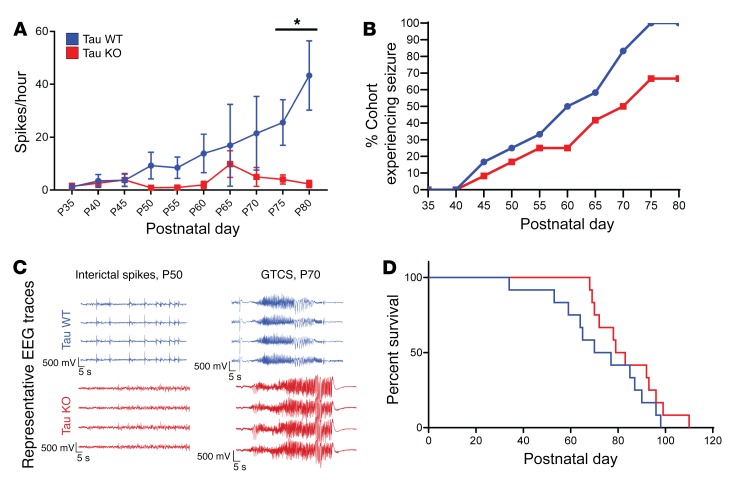
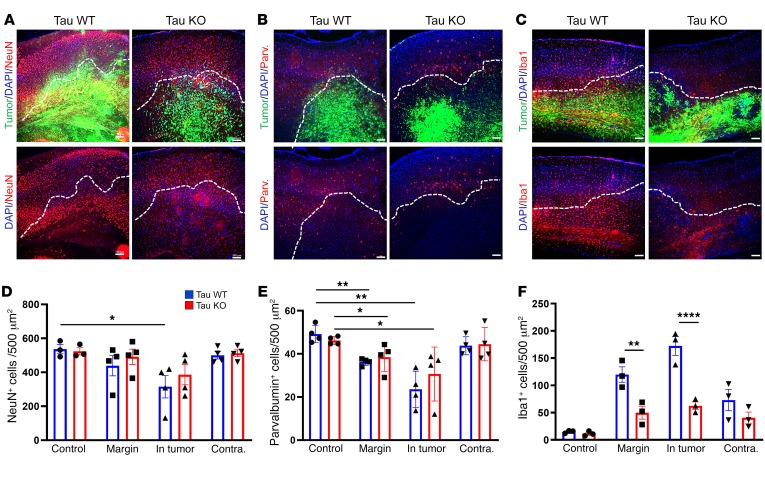
Seizures often herald the clinical appearance of gliomas or appear at later stages. Dissecting their precise evolution and cellular pathogenesis in brain malignancies could inform the development of staged therapies for these highly pharmaco-resistant epilepsies. Studies in immunodeficient xenograft models have identified local interneuron loss and excess glial glutamate release as chief contributors to network disinhibition, but how hyperexcitability in the peritumoral microenvironment evolves in an immunocompetent brain is unclear. We generated gliomas in WT mice via in utero deletion of key tumor suppressor genes and serially monitored cortical epileptogenesis during tumor infiltration with in vivo electrophysiology and GCAMP7 calcium imaging, revealing a reproducible progression from hyperexcitability to convulsive seizures. Long before seizures, coincident with loss of inhibitory cells and their protective scaffolding, gain of glial glutamate antiporter xCT expression, and reactive astrocytosis, we detected local Iba1+ microglial inflammation that intensified and later extended far beyond tumor boundaries. Hitherto unrecognized episodes of cortical spreading depolarization that arose frequently from the peritumoral region may provide a mechanism for transient neurological deficits. Early blockade of glial xCT activity inhibited later seizures, and genomic reduction of host brain excitability by deleting MapT suppressed molecular markers of epileptogenesis and seizures. Our studies confirmed xenograft tumor-driven pathobiology and revealed early and late components of tumor-related epileptogenesis in a genetically tractable, immunocompetent mouse model of glioma, allowing the complex dissection of tumor versus host pathogenic seizure mechanisms.
Keywords: Brain cancer; Epilepsy; Neurological disorders; Oncology; Therapeutics.
Conflict of interest statement
Figures
Comment in
-
Breaking Down the Wall: The Strategic Plan of Cancer to Conquer the Brain.Epilepsy Curr. 2020 Sep 27;20(6):384-386. doi: 10.1177/1535759720949241. eCollection 2020 Nov-Dec. Epilepsy Curr. 2020. PMID: 34025261 Free PMC article. No abstract available.
References
-
- Dührsen L, et al. Seizures as presenting symptom in patients with glioblastoma. Epilepsia. 2019;60(1):149–154. - PubMed
